Basic Spatial Transcriptomics Analysis with Harpy#
In this notebook, we will work with mouse liver spatial transcriptomics data generated using Resolve Biosciences’ Molecular Cartography technology.
import harpy as hp
1. Read in the data#
The first step includes reading in the raw data.
The example dataset for this notebook will be downloaded and cached using pooch via harpy.dataset.registry.
Convert to zarr and read the image#
import os
import tempfile
import uuid
from spatialdata import SpatialData
from spatialdata import read_zarr
from harpy.datasets.registry import get_registry
from spatialdata.transformations import Scale, Identity
from dask_image import imread
# change this path. It is the directory where the spatialdata .zarr will be saved.
OUTPUT_DIR = tempfile.gettempdir()
image_name = "raw_image"
path = None # If None, example data will be downloaded in the default cache folder of your os. Set this to a custom path, to change this behaviour.
registry = get_registry(path=path)
path_image = registry.fetch("transcriptomics/resolve/mouse/20272_slide1_A1-1_DAPI.tiff")
# you can choose any name for your zarr file
output_path = os.path.join(OUTPUT_DIR, f"sdata_{uuid.uuid4()}.zarr")
sdata = SpatialData()
sdata.write(output_path)
sdata = read_zarr(sdata.path)
arr = imread.imread(path_image)
# we will add the image in the micron and pixel coordinate system
pixel_size = 0.138 # pixel size in micron for resolve
scale = Scale(axes=["x", "y"], scale=[pixel_size, pixel_size])
sdata = hp.im.add_image(
sdata,
arr=arr.rechunk(1024),
output_image_name=image_name,
transformations={"pixel": Identity(), "micron": scale},
scale_factors=[2, 2, 2, 2],
c_coords="DAPI",
)
sdata
SpatialData object, with associated Zarr store: /private/var/folders/q5/7yhs0l6d0x771g7qdbhvkvmr0000gp/T/sdata_21e9705d-6c32-4296-b47c-b06327941efa.zarr
└── Images
└── 'raw_image': DataTree[cyx] (1, 12864, 10720), (1, 6432, 5360), (1, 3216, 2680), (1, 1608, 1340), (1, 804, 670)
with coordinate systems:
▸ 'micron', with elements:
raw_image (Images)
▸ 'pixel', with elements:
raw_image (Images)
2. Plot the image#
import numpy as np
import dask.array as da
from matplotlib.colors import Normalize
import matplotlib.pyplot as plt
fig, ax = plt.subplots(1, 1, figsize=(4, 4))
channel = "DAPI"
crd = [3000, 4000, 3000, 4000] # crop is in pixel coordinates
to_coordinate_system = "pixel"
# normalization parameters for visualization (underlying image not changed)
# for visualization we normalize by the 99th percentile
se = hp.im.get_dataarray(sdata, element_name=image_name)
_channel_idx = np.where(se.c.data == channel)[0].item()
vmax = da.percentile(se.data[_channel_idx].flatten(), q=99).compute()
norm = Normalize(vmax=vmax, clip=False)
render_images_kwargs = {"cmap": "viridis", "norm": norm}
show_kwargs = {"title": channel, "colorbar": False}
ax = hp.pl.plot_sdata(
sdata,
image_name=image_name,
channel=channel,
crd=crd,
to_coordinate_system=to_coordinate_system,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=ax,
)
# frameless figure
ax.axis("off")
plt.tight_layout()
plt.show()
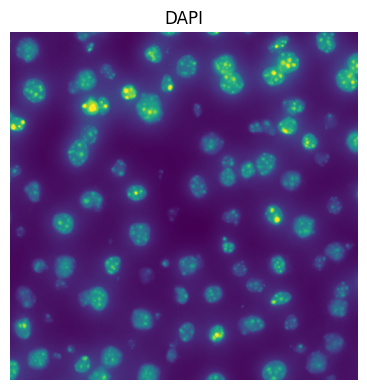
Or plot full image in micron coordinates:
fig, ax = plt.subplots(1, 1, figsize=(6, 6))
channel = "DAPI"
to_coordinate_system = "micron"
render_images_kwargs = {
"cmap": "viridis",
"norm": norm,
}
show_kwargs = {
"title": channel,
"colorbar": False,
}
ax = hp.pl.plot_sdata(
sdata,
image_name=image_name,
channel=channel,
crd=None,
to_coordinate_system=to_coordinate_system,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=ax,
)
plt.tight_layout()
plt.show()
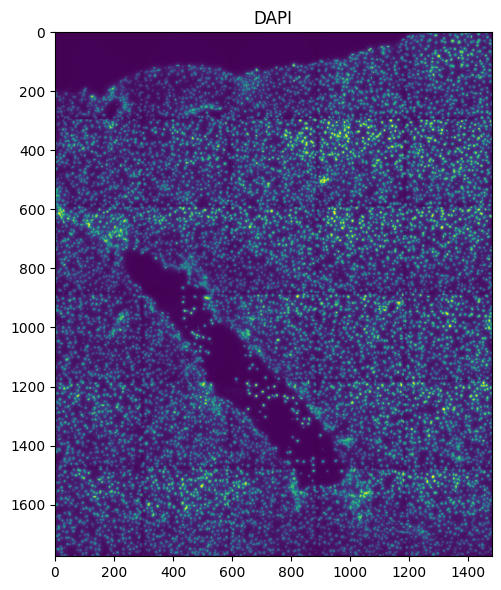
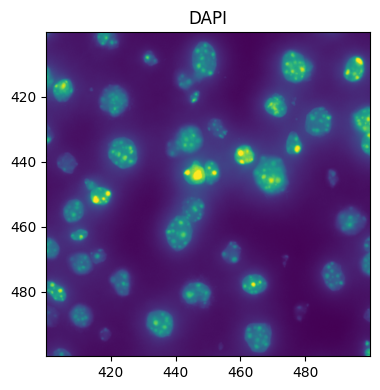
Queries can also be done in micron coordinates:
fig, ax = plt.subplots(1, 1, figsize=(4, 4))
channel = "DAPI"
crd = [400, 500, 400, 500] # crops can also be specified in micron coordinates
to_coordinate_system = "micron"
render_images_kwargs = {"cmap": "viridis", "norm": norm}
show_kwargs = {"title": channel, "colorbar": False}
ax = hp.pl.plot_sdata(
sdata,
image_name=image_name,
channel=channel,
crd=crd,
to_coordinate_system="micron",
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=ax,
)
plt.tight_layout()
plt.show()
3. Segment using Cellpose#
import torch
from dask.distributed import Client, LocalCluster
if torch.cuda.is_available():
from dask_cuda import LocalCUDACluster # pip install dask-cuda
# One worker per GPU; each worker will be pinned to a single GPU.
cluster = LocalCUDACluster(
CUDA_VISIBLE_DEVICES=[0], # or [0,1],...etc
n_workers=1, # 2 if [0,1],...etc
threads_per_worker=1,
memory_limit="32GB",
# local_directory=os.environ.get( "TMPDIR" ),#
)
else:
# cpu/mps fall back
from dask.distributed import LocalCluster
cluster = LocalCluster(
n_workers=1
if torch.backends.mps.is_available()
else 8, # If mps/cuda device available, it is better to increase chunk size to maximal value that fits on the gpu, and set n_workers to 1.
# For this dummy example, we only have one chunk, so setting n_workers>1, has no effect.
threads_per_worker=1,
memory_limit="32GB",
# local_directory=os.environ.get( "TMPDIR" ),
)
client = Client(cluster)
print(client.dashboard_link)
sdata = hp.im.segment(
sdata,
image_name="raw_image",
depth=200,
model=hp.im.cellpose_callable,
# parameters that will be passed to the callable cellpose_callable
diameter=50,
flow_threshold=0.9,
cellprob_threshold=-4,
output_labels_name="segmentation_mask",
output_shapes_name="segmentation_mask_boundaries",
crd=[
3000,
4000,
3000,
4000,
], # region to segment [x_min, xmax, y_min, y_max], must be defined in pixel coordinates
to_coordinate_system="pixel",
overwrite=True,
)
client.close()
http://127.0.0.1:8787/status
4. Visualize resulting segmentation#
fig, axes = plt.subplots(1, 2, figsize=(8, 4))
channel = "DAPI"
image_name = "raw_image"
labels_name = "segmentation_mask"
crd = [
3000,
4000,
3000,
4000,
] # Crop in pixels. One could also choose to do the crop in micron coordinates
to_coordinate_system = "pixel"
render_images_kwargs = {"cmap": "viridis", "norm": norm}
render_labels_kwargs = {"fill_alpha": 0.6, "outline_alpha": 0.4}
show_kwargs = {"title": channel, "colorbar": False}
_ax = hp.pl.plot_sdata(
sdata,
image_name=image_name,
crd=crd,
to_coordinate_system=to_coordinate_system,
channel=channel,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=axes[0],
)
_ax.axis("off")
show_kwargs = {"title": "Segmentation mask", "colorbar": False}
_ax = hp.pl.plot_sdata(
sdata,
image_name=image_name,
labels_name=labels_name,
crd=crd,
to_coordinate_system=to_coordinate_system,
channel=channel,
render_images_kwargs=render_images_kwargs,
render_labels_kwargs=render_labels_kwargs,
show_kwargs=show_kwargs,
ax=axes[1],
)
_ax.axis("off")
plt.tight_layout()
plt.show()
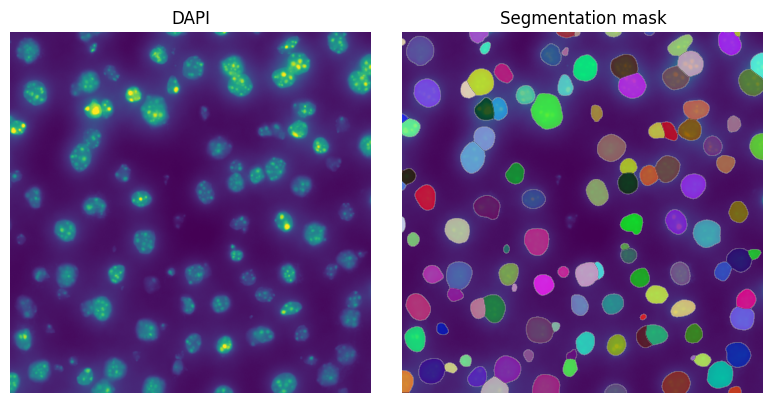
5. Read the transcripts#
path_transcripts = registry.fetch(
"transcriptomics/resolve/mouse/20272_slide1_A1-1_results.txt"
)
sdata = hp.io.read_resolve_transcripts(
sdata,
path_count_matrix=path_transcripts,
output_points_name="transcripts",
to_coordinate_system="pixel",
to_micron_coordinate_system="micron",
pixel_size=pixel_size,
overwrite=True,
)
sdata
SpatialData object, with associated Zarr store: /private/var/folders/q5/7yhs0l6d0x771g7qdbhvkvmr0000gp/T/sdata_21e9705d-6c32-4296-b47c-b06327941efa.zarr
├── Images
│ └── 'raw_image': DataTree[cyx] (1, 12864, 10720), (1, 6432, 5360), (1, 3216, 2680), (1, 1608, 1340), (1, 804, 670)
├── Labels
│ └── 'segmentation_mask': DataArray[yx] (1000, 1000)
├── Points
│ └── 'transcripts': DataFrame with shape: (<Delayed>, 3) (2D points)
└── Shapes
└── 'segmentation_mask_boundaries': GeoDataFrame shape: (121, 1) (2D shapes)
with coordinate systems:
▸ 'micron', with elements:
raw_image (Images), segmentation_mask (Labels), transcripts (Points), segmentation_mask_boundaries (Shapes)
▸ 'pixel', with elements:
raw_image (Images), segmentation_mask (Labels), transcripts (Points), segmentation_mask_boundaries (Shapes)
fig, axes = plt.subplots(1, 2, figsize=(16, 8))
render_images_kwargs = {
"cmap": "binary",
}
show_kwargs = {
"title": "transcripts (in micron)",
"colorbar": False,
}
hp.pl.plot_sdata_genes(
sdata,
points_name="transcripts",
image_name="raw_image",
genes=None, # plot all genes
color="cornflowerblue",
size=0.1,
frac=0.5, # only plot half of them
to_coordinate_system="micron",
show_kwargs=show_kwargs,
render_images_kwargs=render_images_kwargs,
ax=axes[0],
)
render_images_kwargs = {
"cmap": "grey",
}
show_kwargs = {
"title": "transcripts (in micron)",
"colorbar": False,
}
hp.pl.plot_sdata_genes(
sdata,
points_name="transcripts",
image_name="raw_image",
genes=["Axl", "Stab2"],
palette=["#4D88FF", "#FFE082"],
size=5.0,
frac=None,
to_coordinate_system="micron",
show_kwargs=show_kwargs,
render_images_kwargs=render_images_kwargs,
ax=axes[1],
)
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Using 'datashader' backend with 'None' as reduction method to speed up plotting. Depending on the
reduction method, the value range of the plot might change. Set method to 'matplotlib' do disable this
behaviour.
INFO Using 'datashader' backend with 'None' as reduction method to speed up plotting. Depending on the
reduction method, the value range of the plot might change. Set method to 'matplotlib' do disable this
behaviour.
<Axes: title={'center': 'transcripts (in micron)'}>
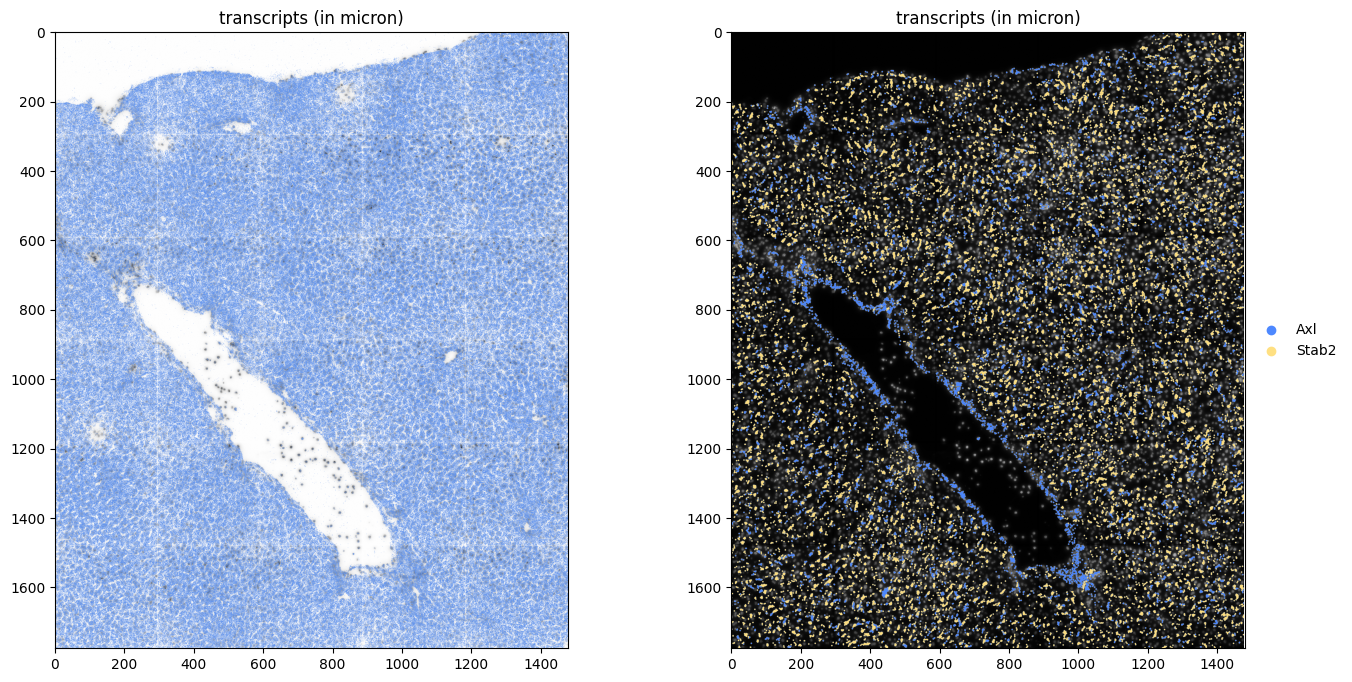
Or visualize a crop.
Querying points is slow in spatialdata, therefore, we query the points outside of spatialdata (and therefore also the image).
from spatialdata import bounding_box_query
crd = [3000, 4000, 3000, 4000]
to_coordinate_system = "pixel" # we query in 'intrinsic' pixel coordinate system
y_start = crd[2]
x_start = crd[0]
y_end = crd[3]
x_end = crd[1]
name_x = "x"
name_y = "y"
y_query = f"{y_start} <={name_y} < {y_end}"
x_query = f"{x_start} <={name_x} < {x_end}"
query = f"{y_query} and {x_query}"
sdata["transcripts"] = sdata["transcripts"].categorize(columns=["gene"])
sdata["transcripts_queried"] = sdata["transcripts"].query(query)
sdata["raw_image_queried"] = bounding_box_query(
sdata["raw_image"],
axes=["x", "y"],
min_coordinate=[crd[0], crd[2]],
max_coordinate=[crd[1], crd[3]],
target_coordinate_system=to_coordinate_system,
)
fig, axes = plt.subplots(1, 1, figsize=(6, 6))
render_images_kwargs = {
"cmap": "pink",
}
show_kwargs = {
"title": channel,
"colorbar": False,
}
hp.pl.plot_sdata_genes(
sdata,
points_name="transcripts_queried",
image_name="raw_image_queried",
genes=["Axl", "Stab2"],
palette=["#4D88FF", "#FFE082"],
size=5.0,
frac=None,
crd=None,
to_coordinate_system=to_coordinate_system,
show_kwargs=show_kwargs,
render_images_kwargs=render_images_kwargs,
ax=axes,
)
del sdata["transcripts_queried"]
del sdata["raw_image_queried"]
axes.axis("off")
(np.float64(3000.0),
np.float64(4000.0),
np.float64(4000.0),
np.float64(3000.0))
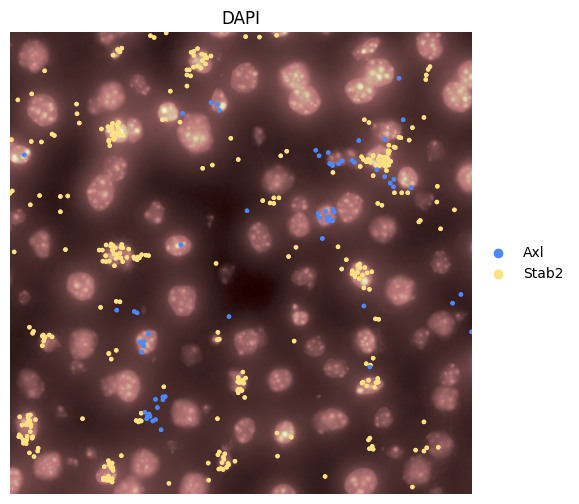
6. Create the AnnData table#
# Use dask cluster for optimal processing. For this dummy example, one could also work without a dask cluster.
cluster = LocalCluster(
n_workers=8,
threads_per_worker=1,
processes=True,
memory_limit="8GB",
# local_directory=os.environ.get( "TMPDIR" ),
)
client = Client(cluster)
print(client.dashboard_link)
http://127.0.0.1:49706/status
sdata = hp.tb.allocate(
sdata,
labels_name="segmentation_mask",
points_name="transcripts",
output_table_name="table_transcriptomics",
chunks=None,
to_coordinate_system="pixel",
update_shapes_elements=False,
overwrite=True,
)
sdata["table_transcriptomics"]
client.close()
sdata["table_transcriptomics"].to_df().head()
| Adamtsl2 | Adgre1 | Atp6v0d2 | Axl | C5ar1 | Ccr2 | Cd207 | Cd209a | Cd36 | Cd3e | ... | Sox9 | Spn | Spon2 | Stab2 | Timd4 | Tmem119 | Vsig4 | Vwf | Wnt2 | Wnt9b | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cells | |||||||||||||||||||||
| 65_segmentation_mask_a6baaa75 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 69_segmentation_mask_a6baaa75 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 71_segmentation_mask_a6baaa75 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 81_segmentation_mask_a6baaa75 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 83_segmentation_mask_a6baaa75 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
5 rows × 71 columns
Now visualize the expression of 2 genes:
fig, axes = plt.subplots(1, 2, figsize=(12, 5))
channel = "DAPI"
image_name = "raw_image"
labels_name = "segmentation_mask"
table_name = "table_transcriptomics"
crd = [3000, 4000, 3000, 4000]
to_coordinate_system = "pixel"
render_images_kwargs = {"cmap": "binary", "norm": norm}
render_labels_kwargs = {"fill_alpha": 0.6, "outline_alpha": 0.4}
# subset because querying of points is slow, and we do not need them in this visualization
sdata_to_plot = sdata.subset(element_names=[image_name, labels_name, table_name])
color = "Axl"
show_kwargs = {
"title": f"{color}",
"colorbar": True,
}
ax = hp.pl.plot_sdata(
sdata_to_plot,
image_name=image_name,
channel=channel,
crd=crd,
to_coordinate_system=to_coordinate_system,
labels_name=labels_name,
table_name=table_name,
color=color,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=axes[0],
)
ax.axis("off")
# color by mean intensity
color = "Stab2"
show_kwargs = {
"title": f"{color}",
"colorbar": True,
}
ax = hp.pl.plot_sdata(
sdata_to_plot,
image_name=image_name,
channel=channel,
crd=crd,
to_coordinate_system=to_coordinate_system,
labels_name=labels_name,
table_name=table_name,
color=color,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=axes[1],
)
ax.axis("off")
plt.tight_layout()
plt.show()
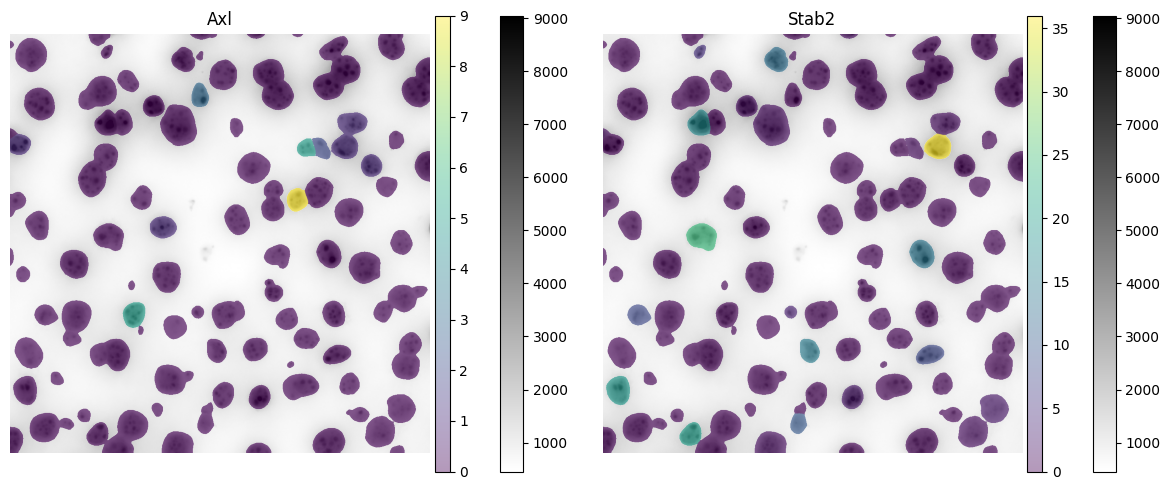
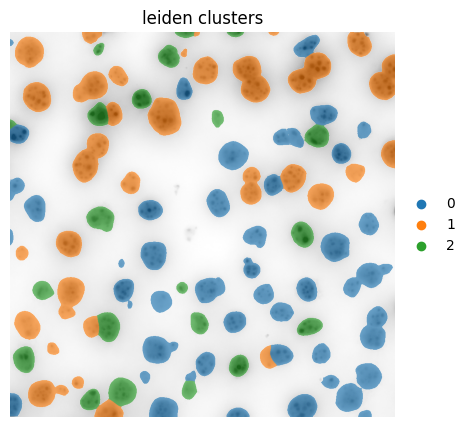
6. Leiden Clustering#
sdata = hp.tb.preprocess_transcriptomics(
sdata,
labels_name="segmentation_mask",
table_name="table_transcriptomics",
output_table_name="table_transcriptomics_preprocessed",
update_shapes_elements=False,
overwrite=True,
)
sdata = hp.tb.leiden(
sdata,
labels_name="segmentation_mask",
table_name="table_transcriptomics_preprocessed",
output_table_name="table_transcriptomics_preprocessed",
overwrite=True,
)
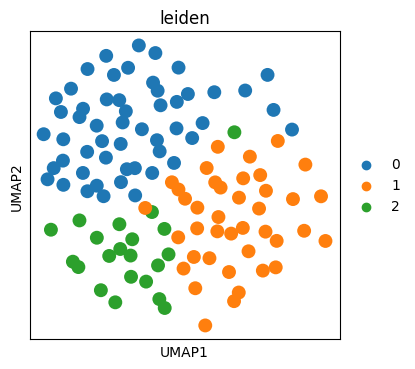
import scanpy as sc
fig, ax = plt.subplots(figsize=(4, 4))
sc.pl.umap(
sdata["table_transcriptomics_preprocessed"],
color="leiden",
size=400,
ax=ax,
)
fig, axes = plt.subplots(1, 1, figsize=(5, 5))
channel = "DAPI"
image_name = "raw_image"
labels_name = "segmentation_mask"
table_name = "table_transcriptomics_preprocessed"
crd = [3000, 4000, 3000, 4000]
to_coordinate_system = "pixel"
render_images_kwargs = {"cmap": "binary", "norm": norm}
render_labels_kwargs = {"fill_alpha": 0.6, "outline_alpha": 0.4}
# subset because querying of points is slow, and we do not need them in this visualization
sdata_to_plot = sdata.subset(element_names=[image_name, labels_name, table_name])
color = "leiden"
show_kwargs = {
"title": f"{color} clusters",
"colorbar": False,
}
ax = hp.pl.plot_sdata(
sdata_to_plot,
image_name=image_name,
channel=channel,
crd=crd,
to_coordinate_system=to_coordinate_system,
labels_name=labels_name,
table_name=table_name,
color=color,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=axes,
)
ax.axis("off")
(np.float64(3000.0),
np.float64(4000.0),
np.float64(4000.0),
np.float64(3000.0))