Run FlowSOM for pixel and cell clustering#
This notebook provides an implementation of unsupervised pixel and cell level clustering approach described in Liu, C.C., Greenwald, N.F., Kong, A., et al. (2023). Robust phenotyping of highly multiplexed tissue imaging data using pixel-level clustering. Nature Communications, 14, 4618. https://doi.org/10.1038/s41467-023-40068-5.
This implementation follows the methodology of the original ark-analysis repository but has been reengineered for compatibility with SpatialData and leverages Dask for efficient scaling to gigapixel-scale images.
You can run this notebook, without installing squidpy, but to run the cell that calls hp.tb.spatial_pixel_neighbors, you’ll need to install it.
import harpy as hp
from harpy.datasets import pixie_example
from harpy.utils._keys import ClusteringKey
# supress warnings
import warnings
warnings.filterwarnings(
"ignore", message="The table is annotating", category=UserWarning
)
1. Load example dataset#
import os
import tempfile
from spatialdata import read_zarr
sdata = pixie_example()
# back the spatialdata object to a zarr store:
OUTPUT_DIR = tempfile.gettempdir()
zarr_path = os.path.join(OUTPUT_DIR, "sdata_pixie.zarr")
sdata.write(zarr_path, overwrite=True)
sdata = read_zarr(sdata.path)
channels = [
"CD3",
"CD4",
"CD8",
"CD14",
"CD20",
"CD31",
"CD45",
"CD68",
"CD163",
"CK17",
"Collagen1",
"Fibronectin",
"ECAD",
"HLADR",
"SMA",
"Vim",
]
import numpy as np
import dask.array as da
from matplotlib.colors import Normalize
import matplotlib.pyplot as plt
fig, axes = plt.subplots(4, 4, figsize=(16, 16))
_image_name = "raw_image_fov0"
to_coordinate_system = "fov0"
for _ax, _channel in zip(axes.flatten(), channels, strict=True):
# normalization parameters for visualization (underlying image not changed)
se = hp.im.get_dataarray(sdata, element_name=_image_name)
_channel_idx = np.where(sdata[_image_name].c.data == _channel)[0].item()
vmax = da.percentile(se.data[_channel_idx].flatten(), q=99).compute()
norm = Normalize(vmax=vmax, clip=False)
render_images_kwargs = {"cmap": "cividis", "norm": norm}
show_kwargs = {"title": _channel, "colorbar": False}
_ax = hp.pl.plot_sdata(
sdata,
image_name=_image_name,
channel=_channel,
to_coordinate_system=to_coordinate_system,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=_ax,
)
# frameless figure
_ax.axis("off")
plt.tight_layout()
plt.show()

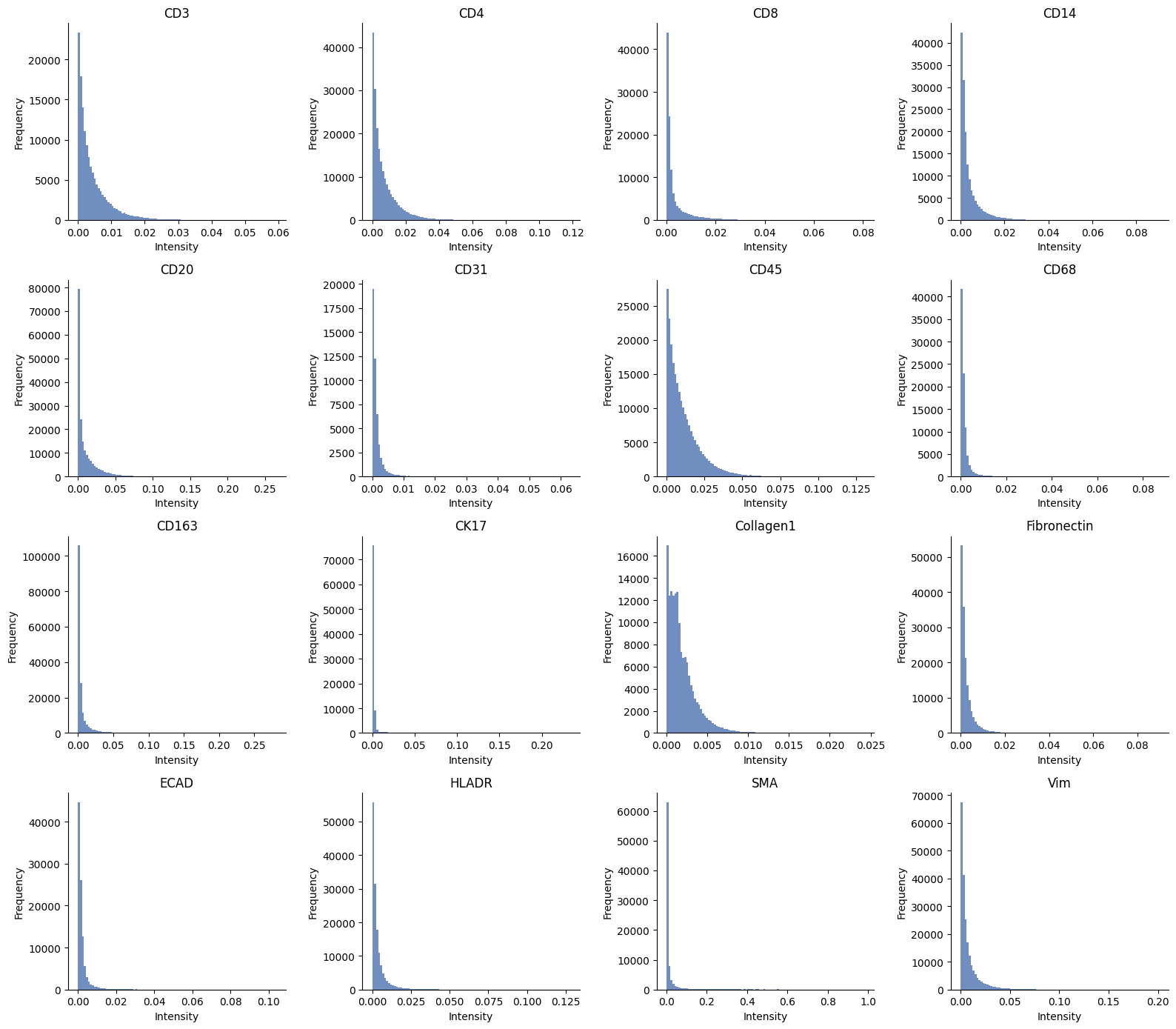
ax = hp.qc.image_histogram(
sdata,
image_name="raw_image_fov0",
channel=channels,
ncols=4,
bins=100,
)
2. Preprocess#
N = 11 # set to 11 to process all samples
image_name = [f"raw_image_fov{i}" for i in range(N)]
sdata = hp.im.pixel_clustering_preprocess(
sdata,
image_name=image_name,
output_image_name=[f"{_image_name}_processed" for _image_name in image_name],
channels=channels,
chunks=2048,
persist_intermediate=True, # set to False if you have multiple images, and if they are large.
overwrite=True,
sigma=2.0,
)
fig, axes = plt.subplots(4, 4, figsize=(16, 16))
_image_name = "raw_image_fov0_processed"
to_coordinate_system = "fov0"
for _ax, _channel in zip(axes.flatten(), channels, strict=True):
render_images_kwargs = {"cmap": "cividis", "norm": None}
show_kwargs = {"title": _channel, "colorbar": False}
_ax = hp.pl.plot_sdata(
sdata,
image_name=_image_name,
channel=_channel,
to_coordinate_system=to_coordinate_system,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=_ax,
)
# frameless figure
_ax.axis("off")
plt.tight_layout()
plt.show()

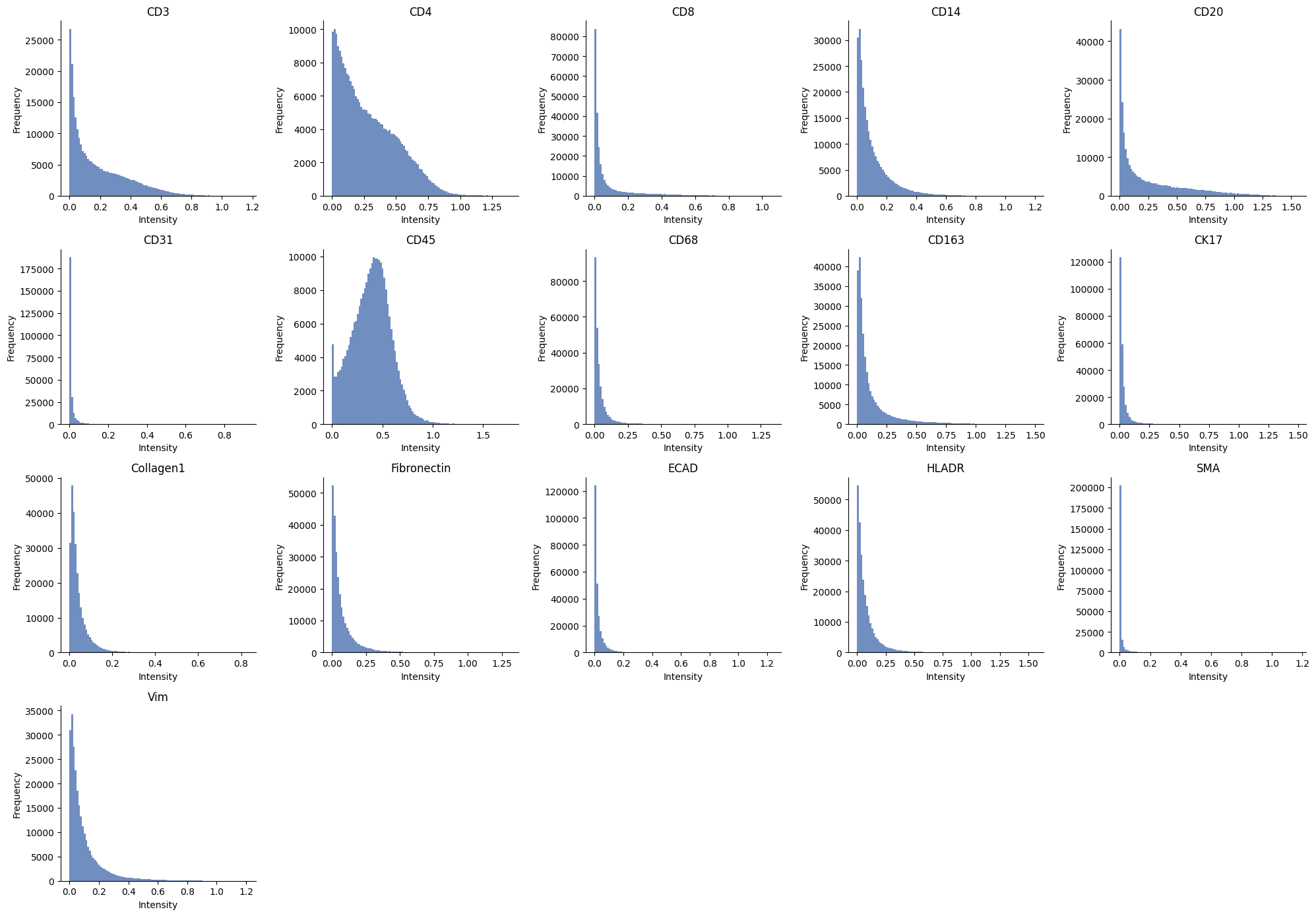
ax = hp.qc.image_histogram(
sdata,
image_name="raw_image_fov0_processed",
channel=channels,
ncols=5,
bins=100,
)
3. Pixel clustering#
import flowsom as fs
from dask.distributed import Client, LocalCluster
work_with_client = True
if work_with_client:
# client example
cluster = LocalCluster(
n_workers=1,
threads_per_worker=10,
)
client = Client(cluster)
print(f"Dashboard link: {client.dashboard_link}")
else:
client = None
batch_model = fs.models.BatchFlowSOMEstimator
sdata, fsom, mapping = hp.im.flowsom(
sdata,
image_name=[f"{_image_name}_processed" for _image_name in image_name],
output_cluster_labels_name=[
f"{_image_name}_flowsom_clusters" for _image_name in image_name
], # we need output_cluster_layer and output_meta_cluster_layer --> these will both be labels elements
output_metacluster_labels_name=[
f"{_image_name}_flowsom_metaclusters" for _image_name in image_name
],
n_clusters=20,
random_state=111,
chunks=512,
client=client,
model=batch_model,
num_batches=10,
xdim=10,
ydim=10,
z_score=True,
z_cap=3,
persist_intermediate=True,
overwrite=True,
)
if client is not None:
client.close()
Dashboard link: http://127.0.0.1:52588/status
sdata = hp.tb.cluster_intensity_SOM(
sdata,
mapping=mapping,
image_name=[f"{_image_name}_processed" for _image_name in image_name],
labels_name=[f"{_image_name}_flowsom_clusters" for _image_name in image_name],
to_coordinate_system=[f"fov{i}" for i in range(N)],
output_table_name="counts_clusters",
overwrite=True,
)
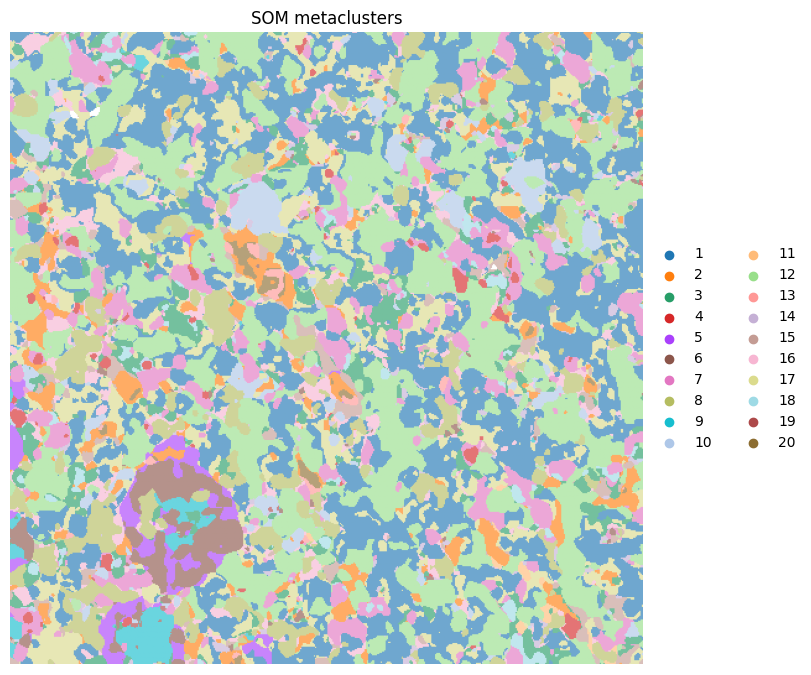
4. Visualization of pixel clusters and metaclusters#
ax = hp.pl.pixel_clusters(
sdata,
labels_name="raw_image_fov0_flowsom_clusters",
figsize=(8, 8),
to_coordinate_system="fov0",
render_labels_kwargs={"alpha": 1},
title="SOM clusters",
)
ax.axis("off")
ax = hp.pl.pixel_clusters(
sdata,
labels_name="raw_image_fov0_flowsom_metaclusters",
figsize=(8, 8),
to_coordinate_system="fov0",
render_labels_kwargs={"alpha": 1},
title="SOM metaclusters",
)
ax.axis("off")
INFO Dropping coordinate system 'fov9' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov10' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov1' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov3' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov7' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov2' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov6' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov5' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov8' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov4' since it doesn't have relevant elements.
INFO Successfully extracted 92 colors from 'cell_ID_cat_colors' in table
'_value_clusters_d442b214-62cd-4feb-b8d5-2182bf62dc84'.
INFO Dropping coordinate system 'fov9' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov10' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov1' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov3' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov7' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov2' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov6' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov5' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov8' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov4' since it doesn't have relevant elements.
INFO Successfully extracted 20 colors from 'cell_ID_cat_colors' in table
'_value_clusters_f33730a1-d402-4580-a627-84de8a59afc6'.
(np.float64(0.0), np.float64(512.0), np.float64(512.0), np.float64(0.0))

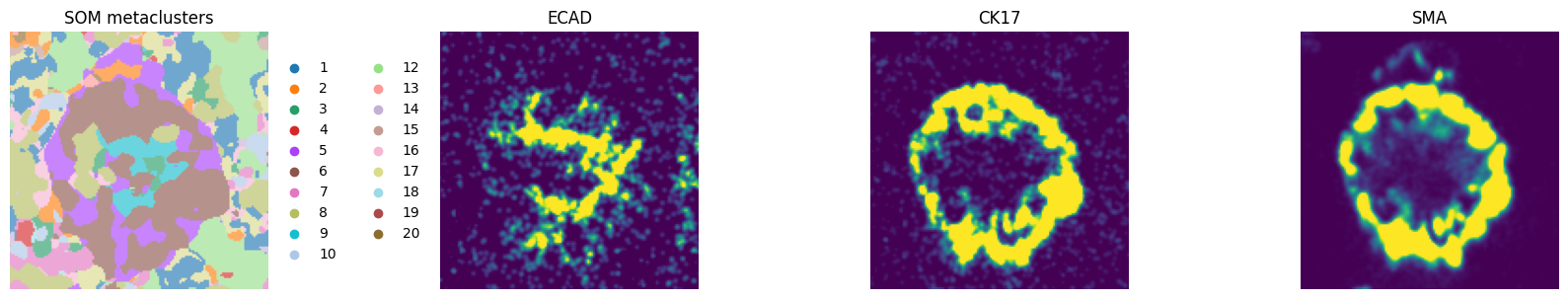
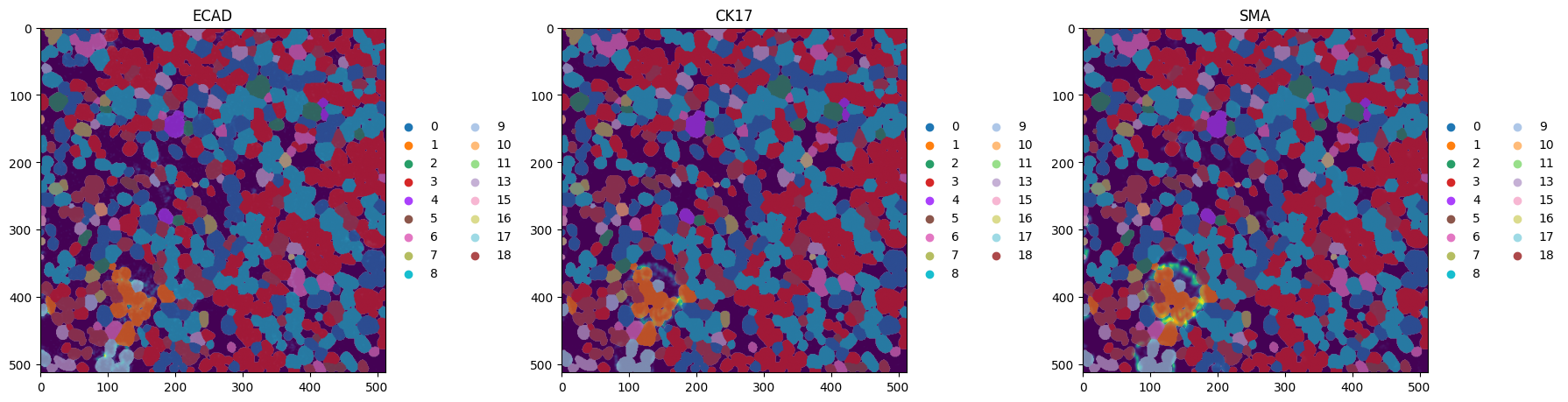
# visualize a crop:
fig, axes = plt.subplots(1, 4, figsize=(16, 4))
crd = [70, 210, 320, 460]
ax = hp.pl.pixel_clusters(
sdata,
labels_name="raw_image_fov0_flowsom_metaclusters",
to_coordinate_system="fov0",
ax=axes[0],
render_labels_kwargs={"alpha": 1},
crd=crd,
title="SOM metaclusters",
)
ax.axis("off")
_image_name = "raw_image_fov0"
subset_channels = [
"ECAD",
"CK17",
"SMA",
]
for _ax, _channel in zip(axes[1:], subset_channels, strict=True):
# normalization parameters for visualization (underlying image not changed)
se = hp.im.get_dataarray(sdata, element_name=_image_name)
_channel_idx = np.where(sdata[_image_name].c.data == _channel)[0].item()
vmax = da.percentile(se.data[_channel_idx].flatten(), q=99).compute()
norm = Normalize(vmax=vmax, clip=False)
render_images_kwargs = {"cmap": "viridis", "norm": norm}
show_kwargs = {"title": _channel, "colorbar": False}
_ax = hp.pl.plot_sdata(
sdata,
image_name=_image_name,
to_coordinate_system="fov0",
channel=_channel,
crd=crd,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=_ax,
)
# frameless figure
_ax.axis("off")
plt.tight_layout()
plt.show()
INFO Dropping coordinate system 'fov9' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov10' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov1' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov3' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov7' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov2' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov6' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov5' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov8' since it doesn't have relevant elements.
INFO Dropping coordinate system 'fov4' since it doesn't have relevant elements.
INFO Successfully extracted 19 colors from 'cell_ID_cat_colors' in table
'_value_clusters_3288e2df-e5b4-48b0-8335-d37f0beb4618'.
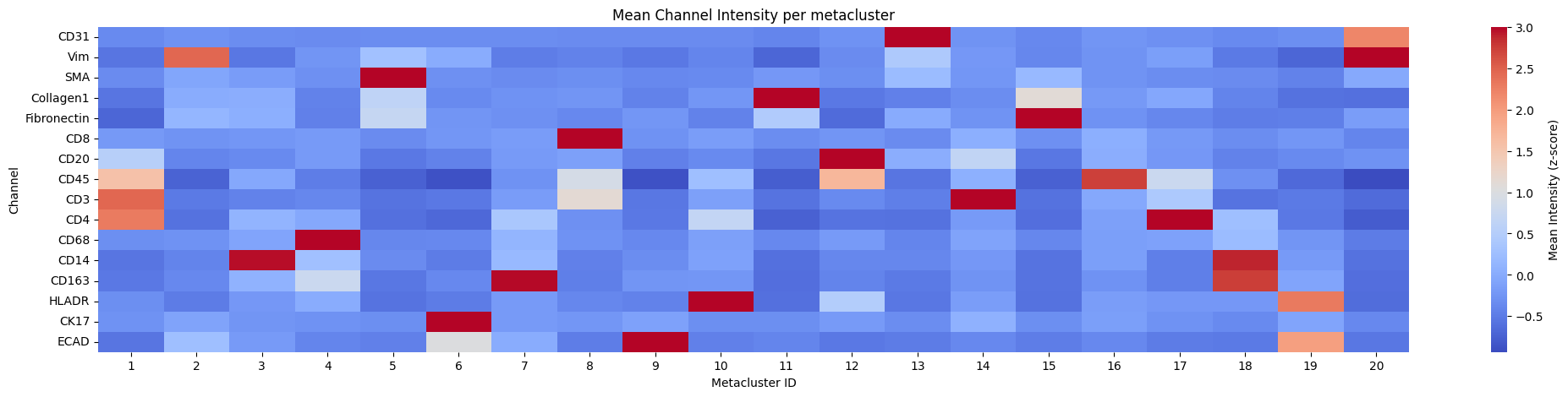
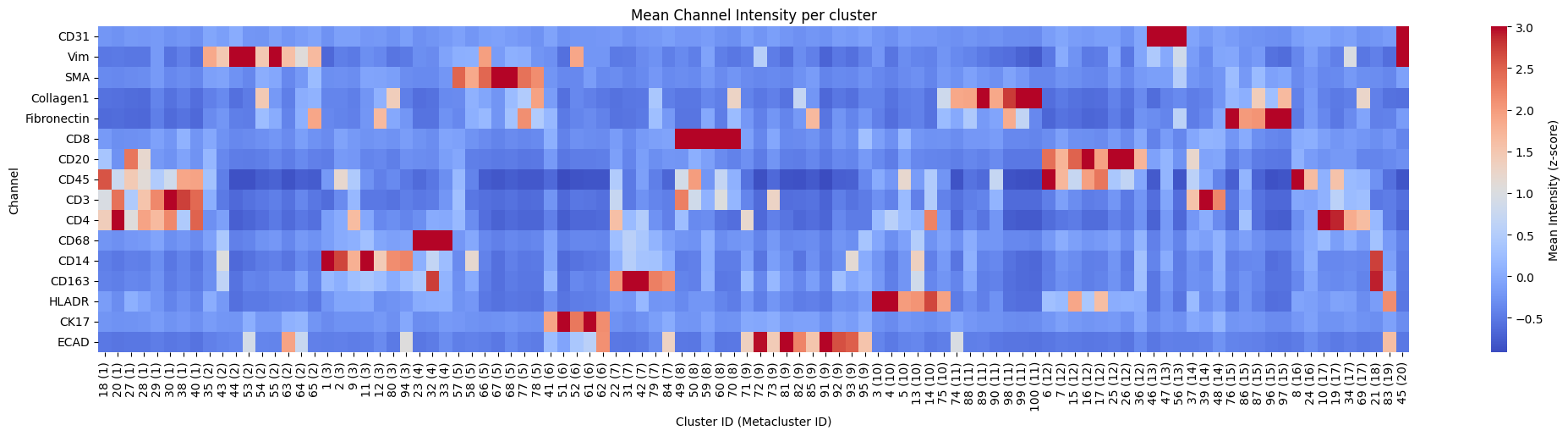
5. Heatmap of channel intensity per cluster and metacluster#
for _metaclusters in [True, False]:
hp.pl.pixel_clusters_heatmap(
sdata,
table_name="counts_clusters",
figsize=(25, 5),
fig_kwargs={"dpi": 100},
linewidths=0.001,
metaclusters=_metaclusters,
z_score=True,
)
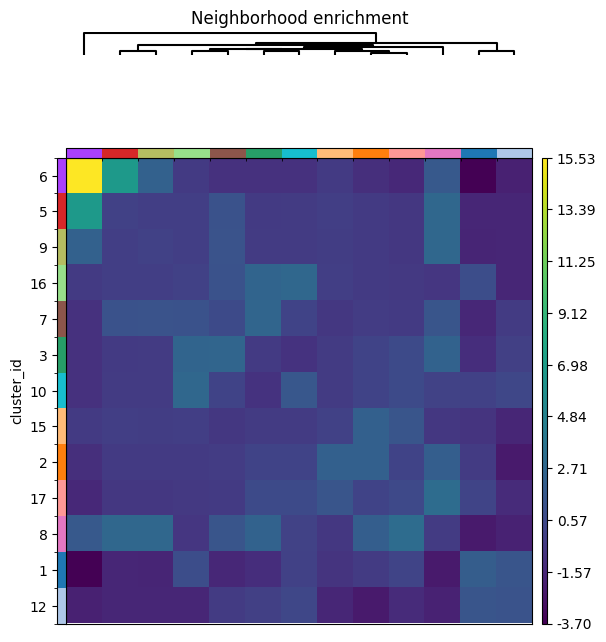
6. Spatial pixel neighbors#
import numpy as np
import squidpy as sq
key_added = "cluster_id"
adata = hp.tb.spatial_pixel_neighbors(
sdata,
labels_name="raw_image_fov0_flowsom_metaclusters",
key_added=key_added,
mode="most_frequent",
grid_type="hexagon",
size=20,
subset=None,
)
adata.uns[f"{key_added}_nhood_enrichment"]["zscore"] = np.nan_to_num(
adata.uns[f"{key_added}_nhood_enrichment"]["zscore"]
)
sq.pl.nhood_enrichment(
adata, cluster_key=key_added, method="ward", mode="zscore", figsize=(6, 6)
)
INFO Creating graph using `grid` coordinates and `None` transform and `1` libraries.
7. Cell clustering#
batch_model = fs.models.BatchFlowSOMEstimator
table_name_flowsom_cell_clustering = "table_cell_clustering_flowsom"
sdata, fsom = hp.tb.flowsom(
sdata,
cells_labels_name=[
f"label_whole_fov{i}" for i in range(N)
], # segmentation masks, can be obtained using hp.im.segment.
cluster_labels_name=[
f"{_image_name}_flowsom_metaclusters" for _image_name in image_name
], # here you could also choose "raw_image_fov0_flowsom_clusters"
output_table_name=table_name_flowsom_cell_clustering,
chunks=512,
model=batch_model,
num_batches=10,
random_state=100,
overwrite=True,
)
Visualize the cell metaclusters:
fig, axes = plt.subplots(1, 3, figsize=(18, 6))
_image_name = "raw_image_fov0"
_labels_name = "label_whole_fov0"
subset_channels = ["ECAD", "CK17", "SMA"]
for _ax, _channel in zip(axes, subset_channels, strict=True):
render_images_kwargs = {"cmap": "viridis", "norm": None}
render_labels_kwargs = {"fill_alpha": 0.4, "outline_alpha": 0.4}
show_kwargs = {"title": _channel, "colorbar": False}
_ax = hp.pl.plot_sdata(
sdata,
image_name=_image_name,
labels_name=_labels_name,
table_name=table_name_flowsom_cell_clustering,
color="metaclustering",
channel=_channel,
to_coordinate_system="fov0",
render_images_kwargs=render_images_kwargs,
render_labels_kwargs=render_labels_kwargs,
show_kwargs=show_kwargs,
ax=_ax,
)
# frameless figure
# _ax.axis("off")
plt.tight_layout()
plt.show()
INFO Successfully extracted 17 colors from 'metaclustering_colors' in table 'table_cell_clustering_flowsom'.
INFO Successfully extracted 17 colors from 'metaclustering_colors' in table 'table_cell_clustering_flowsom'.
INFO Successfully extracted 17 colors from 'metaclustering_colors' in table 'table_cell_clustering_flowsom'.
Now we want to visaulize the mean intensity per cell per cell metacluster, weighted by cell size. We first need to calculate the mean intensities per cell via hp.tb.allocate_intensitiy.
Note that in the ark pipeline, these intensities are weighted by the pixel SOM/META cluster count. To calculate these values, please use the harpy function hp.tb.weighted_channel_expression. Because for regular cell clustering, we only weight the intensities by the size of the cell, we choose to follow the same approach for Flowsom cell clustering.
labels_name = [f"label_whole_fov{i}" for i in range(N)]
to_coordinate_system = [f"fov{i}" for i in range(N)]
table_name = "table_intensities"
for i, (_image_name, _labels_name, _to_coordinate_system) in enumerate(
zip(image_name, labels_name, to_coordinate_system, strict=True)
):
sdata = hp.tb.allocate_intensity(
sdata,
image_name=_image_name,
labels_name=_labels_name,
channels=channels,
output_table_name=table_name,
mode="mean",
to_coordinate_system=_to_coordinate_system,
append=False if i == 0 else True,
overwrite=True,
)
The table element table contains annotated cluster information from the pixie pipeline (Liu, C.C., Greenwald, N.F., Kong, A., et al. (2023)), lets see how they compare to our predicted metaclusters.
We also compare the results with regular leiden clustering on the mean intensity values per cell.
import pandas as pd
from spatialdata.models import TableModel
instance_key = sdata[table_name].uns[TableModel.ATTRS_KEY][TableModel.INSTANCE_KEY]
region_key = sdata[table_name].uns[TableModel.ATTRS_KEY][TableModel.REGION_KEY_KEY]
df = (
sdata["table"]
.obs[["fov", "label", "cell_meta_cluster"]]
.rename(
columns={
"fov": region_key,
"label": instance_key,
"cell_meta_cluster": "cell_type",
}
)
)
index = sdata[table_name].obs.index
sdata[table_name].obs = pd.merge(
sdata[table_name].obs, df, how="inner", on=[instance_key, region_key]
)
sdata[table_name].obs.index = index
Now also add the calculated (via hp.tb.flowsom) cell metacluster and cluster ids to the table element 'table_intensities'.
columns = ["metaclustering", "clustering", instance_key, region_key]
index = sdata[table_name].obs.index
name = sdata[table_name].obs.index.name
sdata[table_name].obs = pd.merge(
sdata[table_name].obs,
sdata[table_name_flowsom_cell_clustering].obs[columns],
on=[instance_key, region_key],
how="left", # left merge, because in table "table_cell_clustering_flowsom", we removed cells with no overlap with any pixel cluster
)
sdata[table_name].obs.index = index
sdata[table_name].obs.index.name = name
# back "table_intensities", now with metaclustering, clustering and cell_type in .obs to zarr
sdata = hp.tb.add_table(
sdata,
adata=sdata[table_name],
output_table_name=table_name,
region=[f"label_whole_fov{i}" for i in range(N)],
overwrite=True,
)
sdata = hp.tb.cluster_intensity(
sdata,
table_name=table_name,
labels_name=[f"label_whole_fov{i}" for i in range(N)],
output_table_name=table_name,
cluster_key="metaclustering",
overwrite=True,
)
ax = hp.pl.cluster_intensity_heatmap(
sdata,
table_name=table_name,
cluster_key="metaclustering",
figsize=(20, 5),
)
ax.set_title("Mean Channel Intensity per cell metacluster")
_x_label = "cell metacluster"
ax.set_ylabel("Channel")
ax.set_xlabel(_x_label)
Text(0.5, 25.722222222222214, 'cell metacluster')
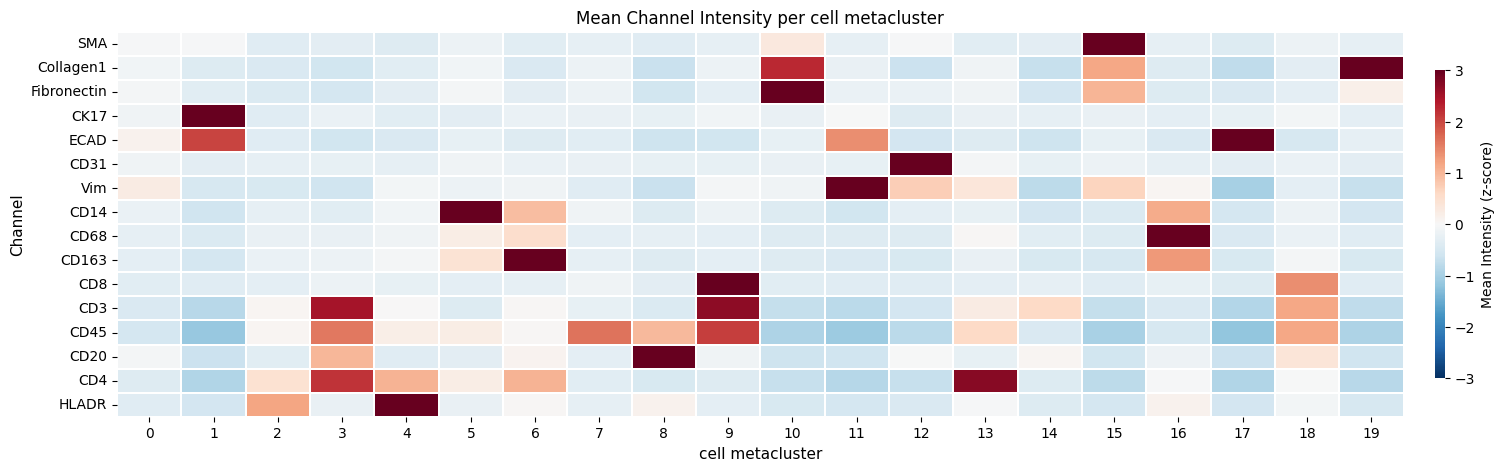
Now compare these results with regular leiden clustering on the mean intensity values.
# normalize before calculating leiden clusters
labels_name = [f"label_whole_fov{i}" for i in range(N)]
sdata = hp.tb.preprocess_proteomics(
sdata,
labels_name=labels_name,
table_name=table_name,
output_table_name=f"{table_name}_normalized",
size_norm=False,
log1p=False,
q=0.99,
max_value_q=1,
overwrite=True,
)
sdata[f"{table_name}_normalized"].to_df().head()
| channels | CD3 | CD4 | CD8 | CD14 | CD20 | CD31 | CD45 | CD68 | CD163 | CK17 | Collagen1 | Fibronectin | ECAD | HLADR | SMA | Vim |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cells | ||||||||||||||||
| 1_label_whole_fov0_09c64416 | 100.000000 | 96.655785 | 9.260165 | 6.321351 | 79.335533 | 0.000000 | 90.805908 | 6.274621 | 0.227555 | 1.503724 | 0.539386 | 1.673003 | 5.863480 | 6.114070 | 0.000000 | 8.542078 |
| 2_label_whole_fov0_09c64416 | 100.000000 | 100.000000 | 1.209254 | 100.000000 | 2.598548 | 1.285120 | 100.000000 | 12.967047 | 16.018177 | 8.001880 | 6.170453 | 0.884670 | 1.455017 | 43.258686 | 0.000000 | 3.866230 |
| 3_label_whole_fov0_09c64416 | 15.790016 | 85.442596 | 1.418018 | 12.311816 | 2.535572 | 1.271602 | 21.691730 | 0.000000 | 6.402584 | 2.490408 | 4.536551 | 6.552049 | 19.387783 | 67.727722 | 1.097208 | 9.684464 |
| 4_label_whole_fov0_09c64416 | 3.287402 | 28.842407 | 1.678827 | 4.460878 | 2.108853 | 0.098956 | 17.441771 | 53.642143 | 7.235740 | 15.314676 | 8.828622 | 6.231126 | 2.147254 | 7.351690 | 1.177836 | 3.420511 |
| 5_label_whole_fov0_09c64416 | 94.864815 | 100.000000 | 1.495612 | 16.172941 | 11.171283 | 2.316715 | 100.000000 | 13.128244 | 10.937386 | 18.826359 | 29.220844 | 22.919373 | 17.487209 | 43.814114 | 7.093751 | 15.714072 |
sdata = hp.tb.leiden(
sdata,
labels_name=labels_name,
table_name=f"{table_name}_normalized",
output_table_name=f"{table_name}_normalized",
key_added="leiden",
overwrite=True,
)
fig, axes = plt.subplots(1, 3, figsize=(18, 6))
_image_name = "raw_image_fov0"
subset_channels = ["ECAD", "CK17", "SMA"]
for _ax, _channel in zip(axes, subset_channels, strict=True):
render_images_kwargs = {"cmap": "viridis", "norm": None}
render_labels_kwargs = {"fill_alpha": 0.4, "outline_alpha": 0.4}
show_kwargs = {"title": _channel, "colorbar": False}
_ax = hp.pl.plot_sdata(
sdata,
image_name=_image_name,
labels_name="label_whole_fov0",
table_name=f"{table_name}_normalized",
color="leiden",
channel=_channel,
to_coordinate_system="fov0",
render_images_kwargs=render_images_kwargs,
render_labels_kwargs=render_labels_kwargs,
show_kwargs=show_kwargs,
ax=_ax,
)
# frameless figure
_ax.axis("off")
plt.tight_layout()
plt.show()
INFO Successfully extracted 11 colors from 'leiden_colors' in table 'table_intensities_normalized'.
INFO Successfully extracted 11 colors from 'leiden_colors' in table 'table_intensities_normalized'.
INFO Successfully extracted 11 colors from 'leiden_colors' in table 'table_intensities_normalized'.
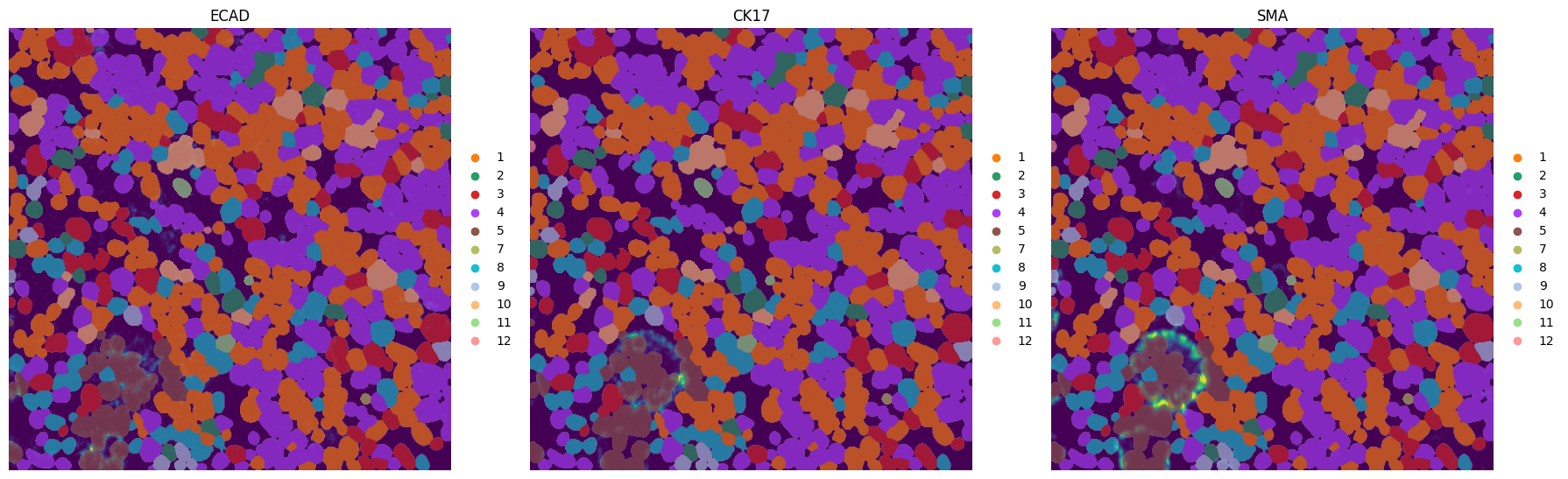
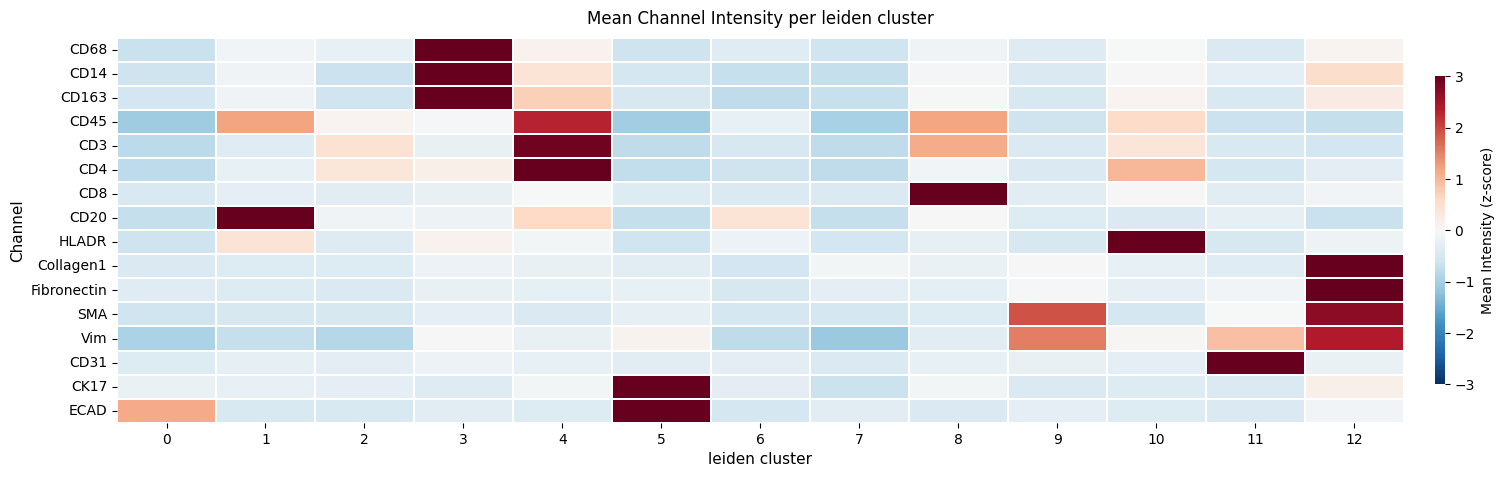
sdata = hp.tb.cluster_intensity(
sdata,
table_name=f"{table_name}_normalized",
labels_name=labels_name,
output_table_name=f"{table_name}_normalized",
cluster_key="leiden",
layer_mean_intensities="raw_counts", # layer in adata.layers that holds the mean intensities
overwrite=True,
)
hp.pl.cluster_intensity_heatmap(
sdata,
table_name=f"{table_name}_normalized",
cluster_key="leiden",
figsize=(20, 5),
)
<Axes: title={'center': 'Mean Channel Intensity per leiden cluster'}, xlabel='leiden cluster', ylabel='Channel'>
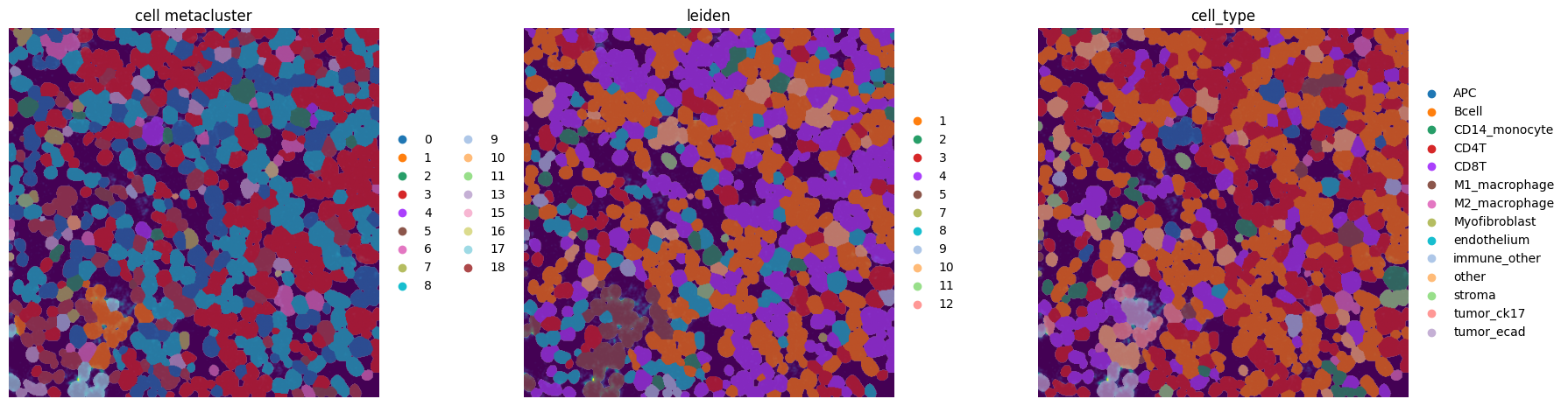
fig, axes = plt.subplots(1, 3, figsize=(18, 6))
_image_name = "raw_image_fov0"
_labels_name = "label_whole_fov0"
_to_coordinate_system = "fov0"
channel = "ECAD"
colors = ["metaclustering", "leiden", "cell_type"]
for _ax, _color in zip(axes, colors, strict=True):
render_images_kwargs = {"cmap": "viridis", "norm": None}
render_labels_kwargs = {"fill_alpha": 0.4, "outline_alpha": 0.4}
show_kwargs = {
"title": "cell metacluster" if _color == "metaclustering" else _color,
"colorbar": False,
}
_ax = hp.pl.plot_sdata(
sdata,
image_name=_image_name,
labels_name=_labels_name,
table_name=f"{table_name}_normalized",
color=_color,
channel=channel,
to_coordinate_system=_to_coordinate_system,
render_images_kwargs=render_images_kwargs,
render_labels_kwargs=render_labels_kwargs,
show_kwargs=show_kwargs,
ax=_ax,
)
# frameless figure
_ax.axis("off")
plt.tight_layout()
plt.show()
INFO Successfully extracted 17 colors from 'metaclustering_colors' in table 'table_intensities_normalized'.
INFO Successfully extracted 11 colors from 'leiden_colors' in table 'table_intensities_normalized'.
INFO Successfully extracted 14 colors from 'cell_type_colors' in table 'table_intensities_normalized'.
import scanpy as sc
# note that these umaps are skwed towards the flowsom metaclusters, because cell type was annotated in Liu, C.C., Greenwald, N.F., Kong, A., et al. (2023) based on the flowsom cell metaclusters.
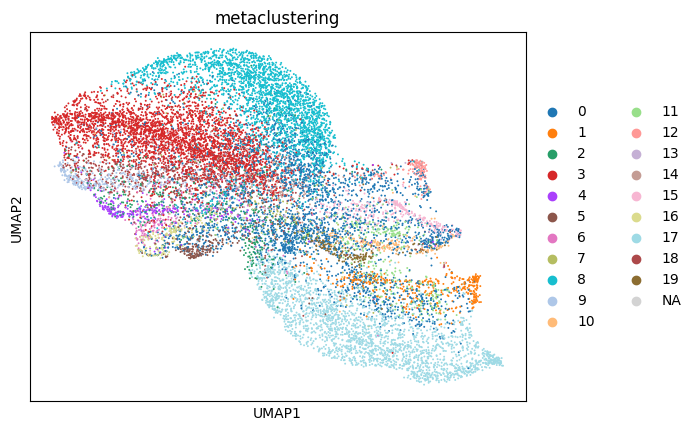
sc.pl.umap(sdata[f"{table_name}_normalized"], color=["metaclustering"])
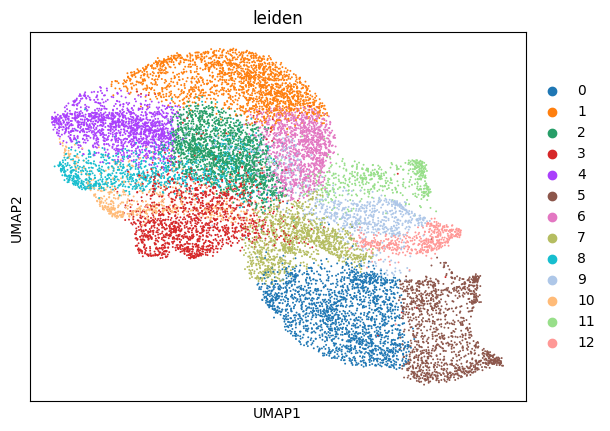
sc.pl.umap(sdata[f"{table_name}_normalized"], color=["leiden"])
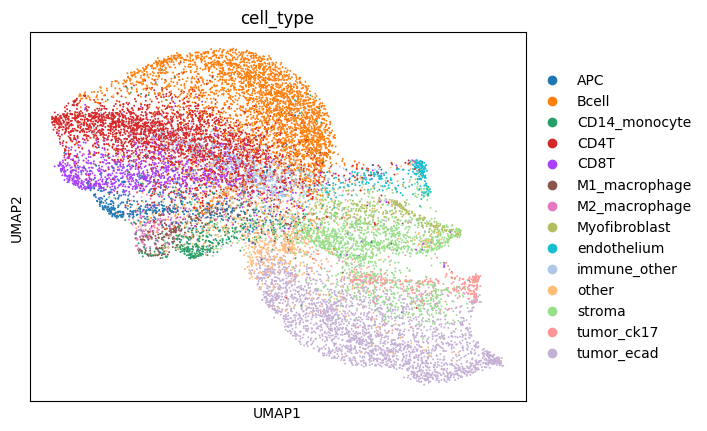
sc.pl.umap(sdata[f"{table_name}_normalized"], color=["cell_type"])
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import seaborn as sns
from scipy.stats import chi2_contingency
table_name = "table_intensities_normalized"
for _cluster_key in ["metaclustering", "leiden"]:
df = sdata[table_name].obs.copy()
df = df[~df["cell_type"].isna()]
contingency = pd.crosstab(df["cell_type"], df[_cluster_key])
# chi squared test
chi2, p, dof, expected = chi2_contingency(contingency)
# cramers v
def _cramers_v(confusion_matrix):
chi2 = chi2_contingency(confusion_matrix)[0]
n = confusion_matrix.sum().sum()
phi2 = chi2 / n
r, k = confusion_matrix.shape
return np.sqrt(phi2 / min(k - 1, r - 1))
print("Cramers v:", _cramers_v(contingency))
print("Chi-squared statistic:", chi2)
print("p-value:", p)
plt.figure(figsize=(10, 5))
sns.heatmap(contingency, cmap="viridis")
plt.title(f"Cell type × {_cluster_key} association")
plt.show()
# Calculate standardized residuals
residuals = (contingency - expected) / np.sqrt(expected)
residuals_df = pd.DataFrame(
residuals, index=contingency.index, columns=contingency.columns
)
resid_long = residuals_df.stack().reset_index()
resid_long.columns = ["cell_type", _cluster_key, "std_residual"]
# Sort by strength of deviation
resid_long["abs_resid"] = resid_long["std_residual"].abs()
top_effects = resid_long.sort_values("abs_resid", ascending=False).head(20)
print(top_effects)
plt.figure(figsize=(10, 5))
sns.heatmap(residuals_df, center=0, cmap="coolwarm", annot=False)
plt.title(f"Standardized residuals: cell_type × {_cluster_key}")
plt.show()
Cramers v: 0.69991741063673
Chi-squared statistic: 97654.5324193182
p-value: 0.0
cell_type metaclustering std_residual abs_resid
155 Myofibroblast 15 103.856744 103.856744
277 tumor_ecad 17 91.604831 91.604831
28 Bcell 8 87.202481 87.202481
4 APC 4 77.543951 77.543951
241 tumor_ck17 1 77.285909 77.285909
45 CD14_monocyte 5 77.060010 77.060010
172 endothelium 12 76.526940 76.526940
98 CD8T 18 75.466500 75.466500
126 M2_macrophage 6 73.204699 73.204699
116 M1_macrophage 16 72.479940 72.479940
63 CD4T 3 64.018750 64.018750
89 CD8T 9 61.189459 61.189459
2 APC 2 59.505149 59.505149
231 stroma 11 48.206569 48.206569
194 immune_other 14 46.467947 46.467947
230 stroma 10 35.677866 35.677866
220 stroma 0 32.002908 32.002908
239 stroma 19 29.632755 29.632755
183 immune_other 3 24.820278 24.820278
263 tumor_ecad 3 -22.402067 22.402067
Cramers v: 0.5964185759950482
Chi-squared statistic: 65462.96455749018
p-value: 0.0
cell_type leiden std_residual abs_resid
60 CD8T 8 95.244502 95.244502
10 APC 10 91.814672 91.814672
115 endothelium 11 78.908615 78.908615
14 Bcell 1 72.854824 72.854824
169 tumor_ecad 0 66.835918 66.835918
43 CD4T 4 60.049037 60.049037
100 Myofibroblast 9 49.568946 49.568946
29 CD14_monocyte 3 48.177373 48.177373
174 tumor_ecad 5 44.836489 44.836489
155 stroma 12 42.461703 42.461703
119 immune_other 2 41.272904 41.272904
19 Bcell 6 39.881712 39.881712
41 CD4T 2 36.845028 36.845028
81 M2_macrophage 3 34.920952 34.920952
161 tumor_ck17 5 34.536849 34.536849
137 other 7 33.313912 33.313912
68 M1_macrophage 3 33.243298 33.243298
150 stroma 7 25.112493 25.112493
152 stroma 9 24.516440 24.516440
13 Bcell 0 -19.195630 19.195630
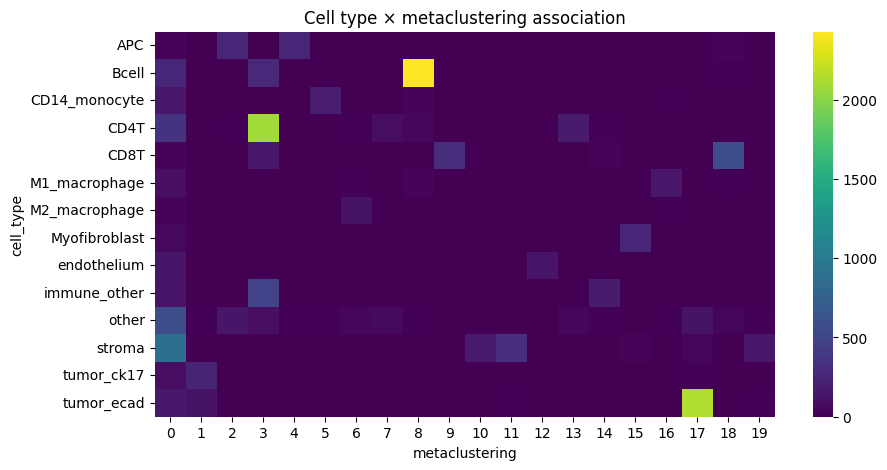
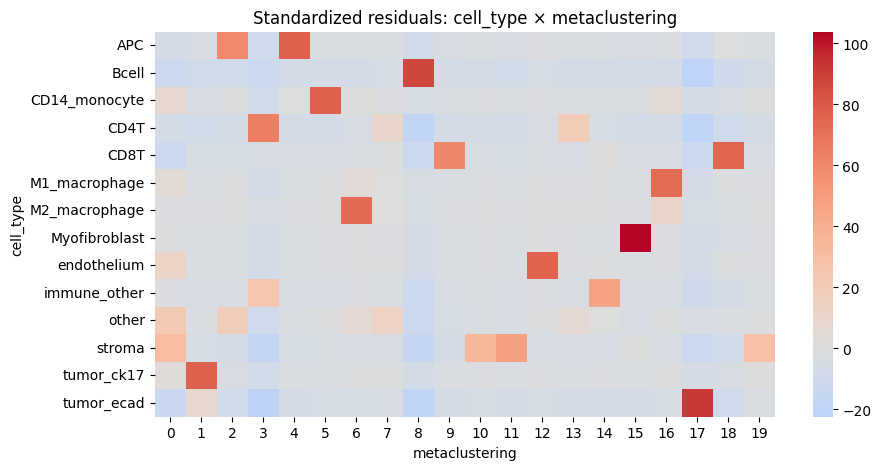
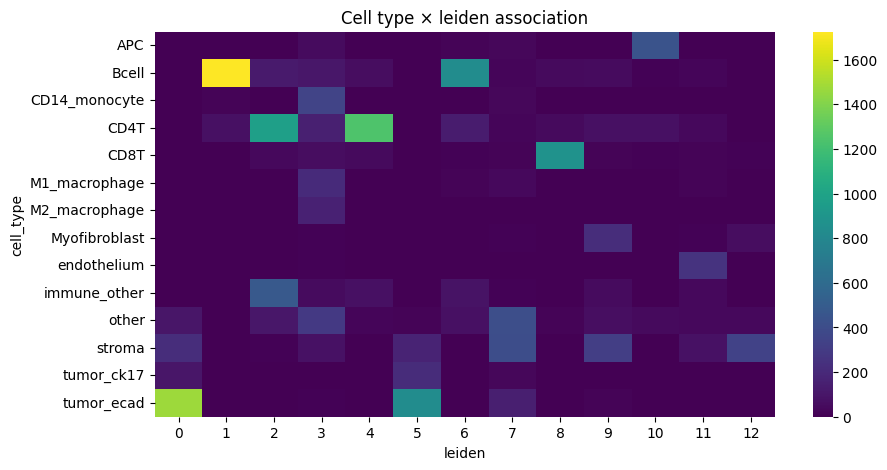
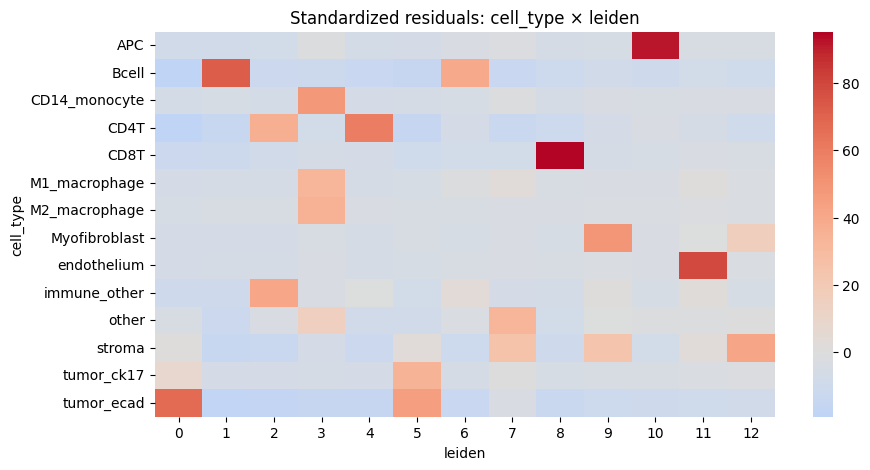
We notice a higher correlation between cell type predicted via cell metaclustering, compared to regular leiden clustering.
# "table_cell_clustering_flowsom" is annotated by segmentation masks, so they can also be visualised using napari-spatialdata
from spatialdata.models import TableModel
sdata["table_cell_clustering_flowsom"].uns[TableModel.ATTRS_KEY]
# from napari_spatialdata import Interactive
# Interactive(sdata)
{'region': ['label_whole_fov0',
'label_whole_fov1',
'label_whole_fov2',
'label_whole_fov3',
'label_whole_fov4',
'label_whole_fov5',
'label_whole_fov6',
'label_whole_fov7',
'label_whole_fov8',
'label_whole_fov9',
'label_whole_fov10'],
'instance_key': 'cell_ID',
'region_key': 'fov_labels'}
Optional export to a .csv format that can be used for visualization using the ark analysis gui#
# weighted channel average for visualization in the ark analysis gui (optional) -> calculate this on the flowsom clustered matrix
sdata = hp.tb.weighted_channel_expression(
sdata,
cell_clustering_table_name="table_cell_clustering_flowsom",
table_name_pixel_cluster_intensity="counts_clusters",
output_table_name="table_cell_clustering_flowsom",
clustering_key=ClusteringKey._METACLUSTERING_KEY,
overwrite=True,
)
from harpy.table.cell_clustering._utils import (
_export_to_ark_format as _export_to_ark_format_cells,
)
from harpy.table.pixel_clustering._cluster_intensity import (
_export_to_ark_format as _export_to_ark_format_pixels,
)
df = _export_to_ark_format_pixels(adata=sdata["counts_clusters"], output=None)
(
df_cell_som_cluster_count_avg,
df_cell_som_cluster_channel_avg,
df_cell_meta_cluster_channel_avg,
) = _export_to_ark_format_cells(
sdata, table_name="table_cell_clustering_flowsom", output=None
)
df.head()
| channels | CD3 | CD4 | CD8 | CD14 | CD20 | CD31 | CD45 | CD68 | CD163 | CK17 | Collagen1 | Fibronectin | ECAD | HLADR | SMA | Vim | pixel_meta_cluster | pixel_som_cluster | count |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SOM_cluster_ID_index | |||||||||||||||||||
| 1_counts_clusters_4a0295d3 | 3.170556 | 12.888708 | 2.673701 | 102.297302 | 1.981421 | 1.010174 | 15.915800 | 5.115088 | 13.101303 | 1.311709 | 7.427465 | 6.707138 | 3.581628 | 7.404450 | 2.121363 | 3.902526 | 3 | 1 | 61858.0 |
| 2_counts_clusters_4a0295d3 | 7.802601 | 15.197968 | 5.868374 | 52.094273 | 6.514609 | 2.945678 | 43.575554 | 6.603859 | 11.311961 | 2.333262 | 7.480127 | 5.921785 | 3.862716 | 10.003304 | 2.255558 | 7.188983 | 3 | 2 | 76777.0 |
| 3_counts_clusters_4a0295d3 | 8.591969 | 18.529282 | 5.463168 | 7.904346 | 4.000842 | 1.638508 | 19.988863 | 6.152431 | 7.790992 | 2.215147 | 7.164235 | 5.330628 | 4.973933 | 78.794853 | 1.134772 | 11.190743 | 10 | 3 | 101431.0 |
| 4_counts_clusters_4a0295d3 | 6.426503 | 24.199673 | 3.257826 | 4.316765 | 1.848322 | 0.914533 | 15.558946 | 3.096722 | 4.072947 | 1.105101 | 4.323403 | 2.858347 | 2.538521 | 117.102592 | 0.420902 | 5.771106 | 10 | 4 | 59752.0 |
| 5_counts_clusters_4a0295d3 | 16.677263 | 19.242825 | 9.086171 | 8.071563 | 7.421441 | 2.405537 | 43.708309 | 5.483037 | 7.129220 | 3.141083 | 6.394245 | 4.868136 | 4.656443 | 43.941631 | 1.605118 | 9.540346 | 10 | 5 | 85247.0 |
df_cell_meta_cluster_channel_avg.head()
| cell_meta_cluster | CD3 | CD4 | CD8 | CD14 | CD20 | CD31 | CD45 | CD68 | CD163 | CK17 | Collagen1 | Fibronectin | ECAD | HLADR | SMA | Vim | cell_meta_cluster_rename | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 13.312110 | 14.540046 | 5.212882 | 10.652877 | 9.342804 | 4.176129 | 19.527418 | 5.825487 | 8.925391 | 4.812573 | 13.868835 | 12.002700 | 10.981284 | 8.484389 | 5.987512 | 19.682054 | 1 |
| 1 | 2 | 6.822442 | 6.668731 | 4.851417 | 7.685502 | 3.745090 | 2.177237 | 6.608780 | 3.866220 | 8.094399 | 32.722800 | 11.086600 | 10.109598 | 32.357767 | 5.390016 | 3.112652 | 15.338476 | 2 |
| 2 | 3 | 15.606988 | 23.245666 | 5.825461 | 10.316361 | 6.800073 | 2.170474 | 24.766719 | 6.335626 | 9.973863 | 3.105719 | 10.342049 | 7.480709 | 9.202182 | 33.578242 | 2.408904 | 10.282013 | 3 |
| 3 | 4 | 40.002591 | 39.067493 | 6.690993 | 6.638652 | 13.699744 | 2.078078 | 41.266476 | 4.937002 | 7.077583 | 3.187016 | 6.369957 | 4.418863 | 3.123610 | 8.655178 | 2.144527 | 8.340994 | 4 |
| 4 | 5 | 12.083061 | 23.661401 | 5.614998 | 11.034528 | 4.662411 | 1.841157 | 24.230448 | 6.375871 | 9.522896 | 2.408798 | 10.240973 | 7.033974 | 4.482985 | 54.909245 | 1.879081 | 9.668706 | 5 |