Example spatial transcriptomics (Xenium)#
Data downloaded from 10x Genomics (accessed 25/11/2025): https://www.10xgenomics.com/datasets/xenium-prime-ffpe-human-ovarian-cancer
1. Read in the data#
import os
import harpy as hp
path_sdata = os.path.join(
os.environ.get("TMPDIR"), "sdata.zarr"
) # or pick a path where the SpatialData object will be backed.
# takes around 7 minutes
sdata = hp.datasets.xenium_human_ovarian_cancer(output=path_sdata)
2. Plot the images#
H&E and annotated H&E
import matplotlib.pyplot as plt
fig, axes = plt.subplots(1, 2, figsize=(20, 10))
image_name = ["he_image_global_ROI1", "he_image_annotated_global_ROI1"]
for _image_name, _ax in zip(image_name, axes, strict=True):
show_kwargs = {
"title": _image_name,
"colorbar": False,
}
ax = hp.pl.plot_sdata(
sdata,
image_name=_image_name,
channel=["r", "g", "b"],
to_coordinate_system="global_ROI1", # or if you want to plot in micron coordinates, specify "global_ROI1_micron"
show_kwargs=show_kwargs,
ax=_ax,
)
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
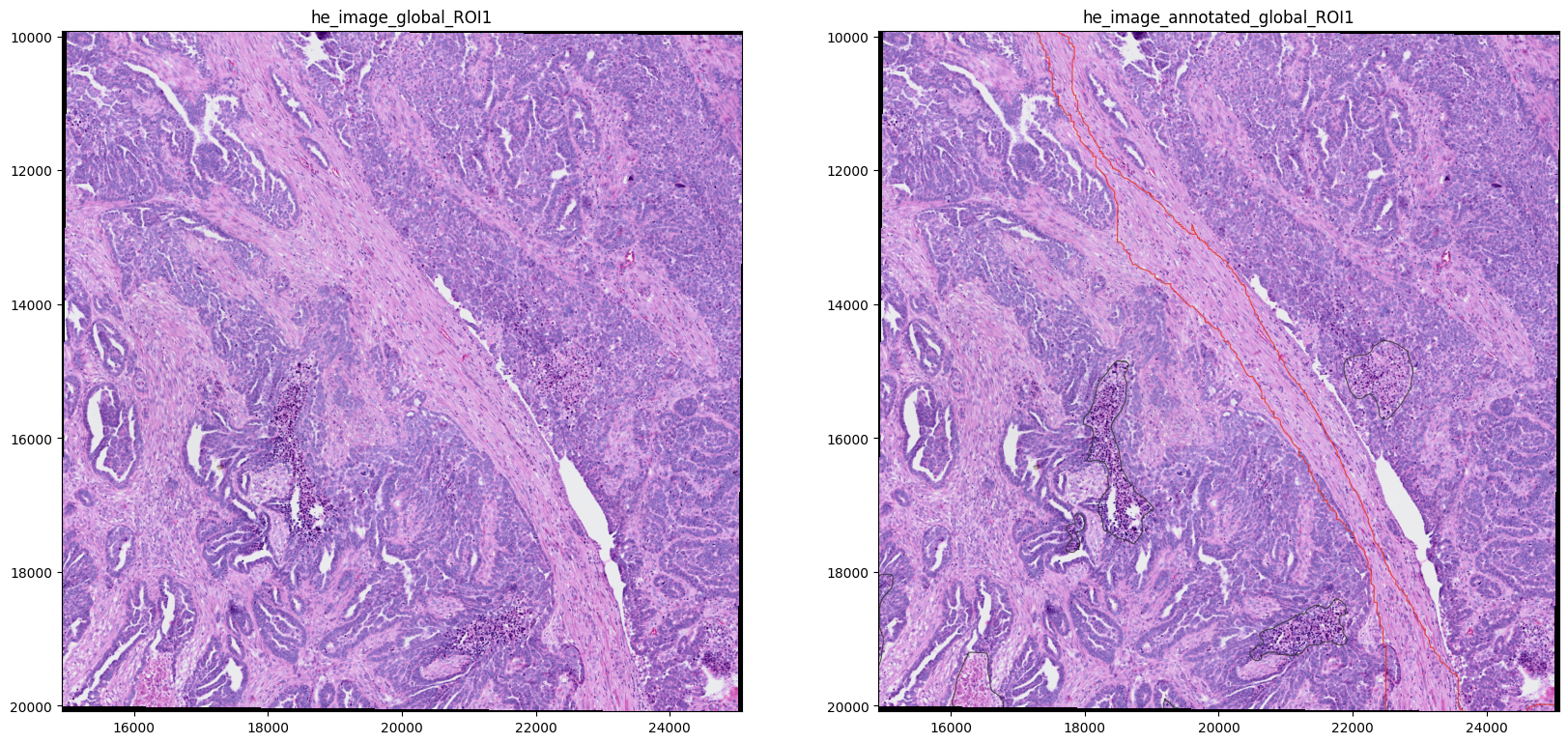
import matplotlib.pyplot as plt
fig, axes = plt.subplots(1, 2, figsize=(20, 10))
show_kwargs = {"colorbar": False}
image_name = ["he_image_global_ROI1", "he_image_annotated_global_ROI1"]
for _image_name, _ax in zip(image_name, axes, strict=True):
show_kwargs = {
"title": _image_name,
"colorbar": False,
}
ax = hp.pl.plot_sdata(
sdata,
image_name=_image_name,
crd=[15000, 25000, 10000, 20000],
channel=["r", "g", "b"],
to_coordinate_system="global_ROI1",
show_kwargs=show_kwargs,
ax=_ax,
)
Morphology focus
This is the image that is used for segmentation.
import matplotlib.pyplot as plt
fig, ax = plt.subplots(1, 1, figsize=(10, 10))
image_name = "morphology_focus_global_ROI1"
channel = hp.im.get_dataarray(sdata, element_name=image_name).c.data.tolist()
show_kwargs = {
"title": image_name,
"colorbar": False,
}
ax = hp.pl.plot_sdata(
sdata,
image_name=image_name,
channel=channel,
to_coordinate_system="global_ROI1", # or if you want to plot in micron coordinates, specify "global_ROI1_micron"
show_kwargs=show_kwargs,
ax=ax,
)
INFO Rasterizing image for faster rendering.
INFO Your image has 4 channels. Sampling categorical colors and using multichannel strategy 'stack' to render.

2. Visualize the transcripts#
import matplotlib.pyplot as plt
fig, ax = plt.subplots(1, 1, figsize=(8, 8))
render_images_kwargs = {
"cmap": "binary",
}
show_kwargs = {
"title": "transcripts",
"colorbar": False,
}
hp.pl.plot_sdata_genes(
sdata,
points_name="transcripts_global_ROI1",
image_name="morphology_focus_global_ROI1",
channel="DAPI",
genes=None, # plot all genes
color="cornflowerblue",
size=0.1,
frac=0.01, # only plot 1% of them
to_coordinate_system="global_ROI1",
show_kwargs=show_kwargs,
render_images_kwargs=render_images_kwargs,
ax=ax,
)
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Rasterizing image for faster rendering.
INFO Using 'datashader' backend with 'None' as reduction method to speed up plotting. Depending on the
reduction method, the value range of the plot might change. Set method to 'matplotlib' do disable this
behaviour.
<Axes: title={'center': 'transcripts'}>

Now plot some genes associated with tumor cells.
import matplotlib.pyplot as plt
fig, axes = plt.subplots(1, 2, figsize=(16, 8))
render_images_kwargs = {
"cmap": "binary",
}
for gene, ax in zip(["PLXNB1", "CP"], axes, strict=True):
show_kwargs = {
"title": gene,
"colorbar": False,
}
hp.pl.plot_sdata_genes(
sdata,
points_name="transcripts_global_ROI1",
image_name="morphology_focus_global_ROI1",
channel="DAPI",
genes=[
gene
], # You could specify a list of genes here. But note that if they occur at same location, they will plotted onto each other.
palette=["#4D88FF"],
size=0.1,
frac=None,
to_coordinate_system="global_ROI1",
show_kwargs=show_kwargs,
render_images_kwargs=render_images_kwargs,
ax=ax,
)
plt.tight_layout()
plt.show()
INFO Rasterizing image for faster rendering.
INFO input has more than 103 categories. Uniform 'grey' color will be used for all categories.
INFO Using 'datashader' backend with 'None' as reduction method to speed up plotting. Depending on the
reduction method, the value range of the plot might change. Set method to 'matplotlib' do disable this
behaviour.
INFO Rasterizing image for faster rendering.
INFO input has more than 103 categories. Uniform 'grey' color will be used for all categories.
INFO Using 'datashader' backend with 'None' as reduction method to speed up plotting. Depending on the
reduction method, the value range of the plot might change. Set method to 'matplotlib' do disable this
behaviour.
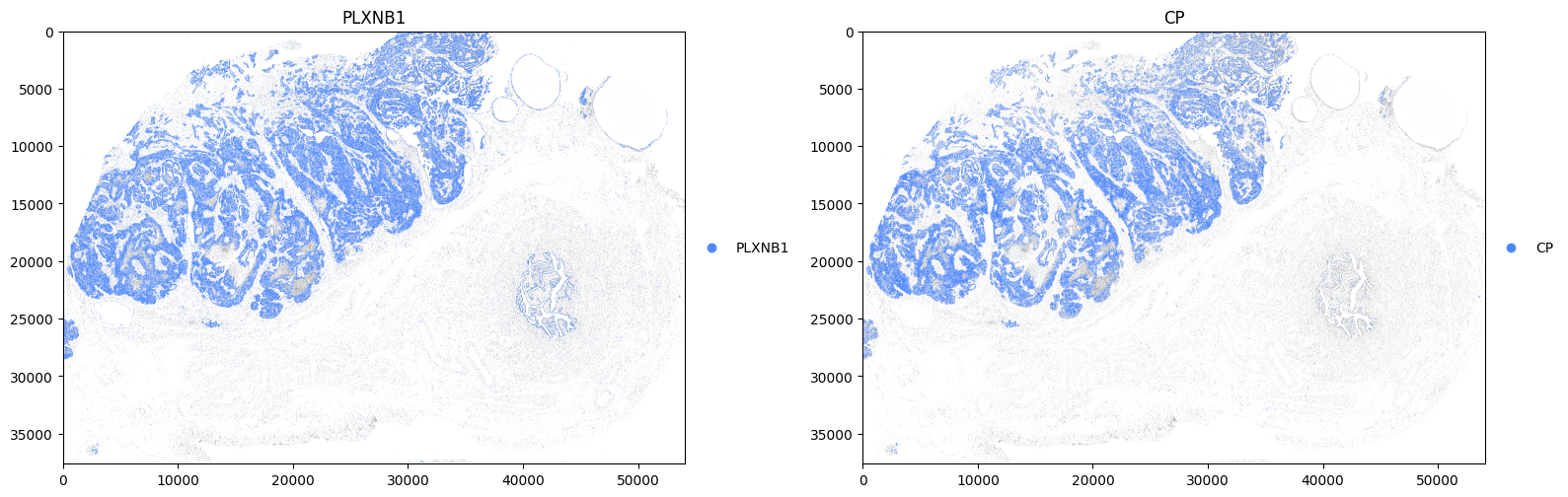
Calculate the transcript density
sdata = hp.im.transcript_density(
sdata,
points_name="transcripts_global_ROI1",
image_name="morphology_focus_global_ROI1",
output_image_name="transcript_density",
to_coordinate_system="global_ROI1",
scale_factors=[2, 2, 2, 2],
overwrite=True,
)
import numpy as np
import dask.array as da
from matplotlib.colors import Normalize
scale = None # auto pick scale by spatialdata-plot
image_name = "transcript_density"
fig, ax = plt.subplots(1, 1, figsize=(10, 10))
channel = 0
se = hp.im.get_dataarray(sdata, element_name=image_name)
_channel_idx = np.where(se.c.data == channel)[0].item()
vmax = da.percentile(se.data[_channel_idx].flatten(), q=95).compute()
norm = Normalize(vmax=vmax, clip=False)
print(norm)
crd = None
sdata_to_plot = sdata.subset(element_names=[image_name])
render_images_kwargs = {"cmap": "grey", "scale": scale, "norm": norm}
show_kwargs = {
"title": "transcript density",
"colorbar": False,
}
ax = hp.pl.plot_sdata(
sdata_to_plot,
image_name=image_name,
channel=channel,
crd=crd,
to_coordinate_system="global_ROI1",
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=ax,
)
plt.tight_layout()
plt.show()
<matplotlib.colors.Normalize object at 0x7f4e992eae40>
INFO Rasterizing image for faster rendering.

3. Segmentation#
Segmentation is already provided by 10x Genomics. We will inspect these segmentation masks, and also illustate how the user could generate segmentation masks using harpy.
First visualize segmentation masks provided by 10x Genomics.
fig, axes = plt.subplots(1, 2, figsize=(20, 10))
channel = "DAPI"
image_name = "morphology_focus_global_ROI1"
labels_name = "cell_labels_global_ROI1"
crd = [15000, 20000, 10000, 15000]
to_coordinate_system = "global_ROI1"
render_images_kwargs = {"cmap": "viridis"}
render_labels_kwargs = {"fill_alpha": 0.6, "outline_alpha": 0.4}
show_kwargs = {"title": f"DAPI in {to_coordinate_system}", "colorbar": False}
hp.pl.plot_sdata(
sdata,
image_name=image_name,
crd=crd,
to_coordinate_system=to_coordinate_system,
channel=channel,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=axes[0],
)
# _ax.axis("off")
show_kwargs = {
"title": f"Segmentation mask in {to_coordinate_system}",
"colorbar": False,
}
hp.pl.plot_sdata(
sdata,
image_name=image_name,
labels_name=labels_name,
crd=crd,
to_coordinate_system=to_coordinate_system,
channel=channel,
render_images_kwargs=render_images_kwargs,
render_labels_kwargs=render_labels_kwargs,
show_kwargs=show_kwargs,
ax=axes[1],
)
# _ax.axis("off")
plt.tight_layout()
plt.show()
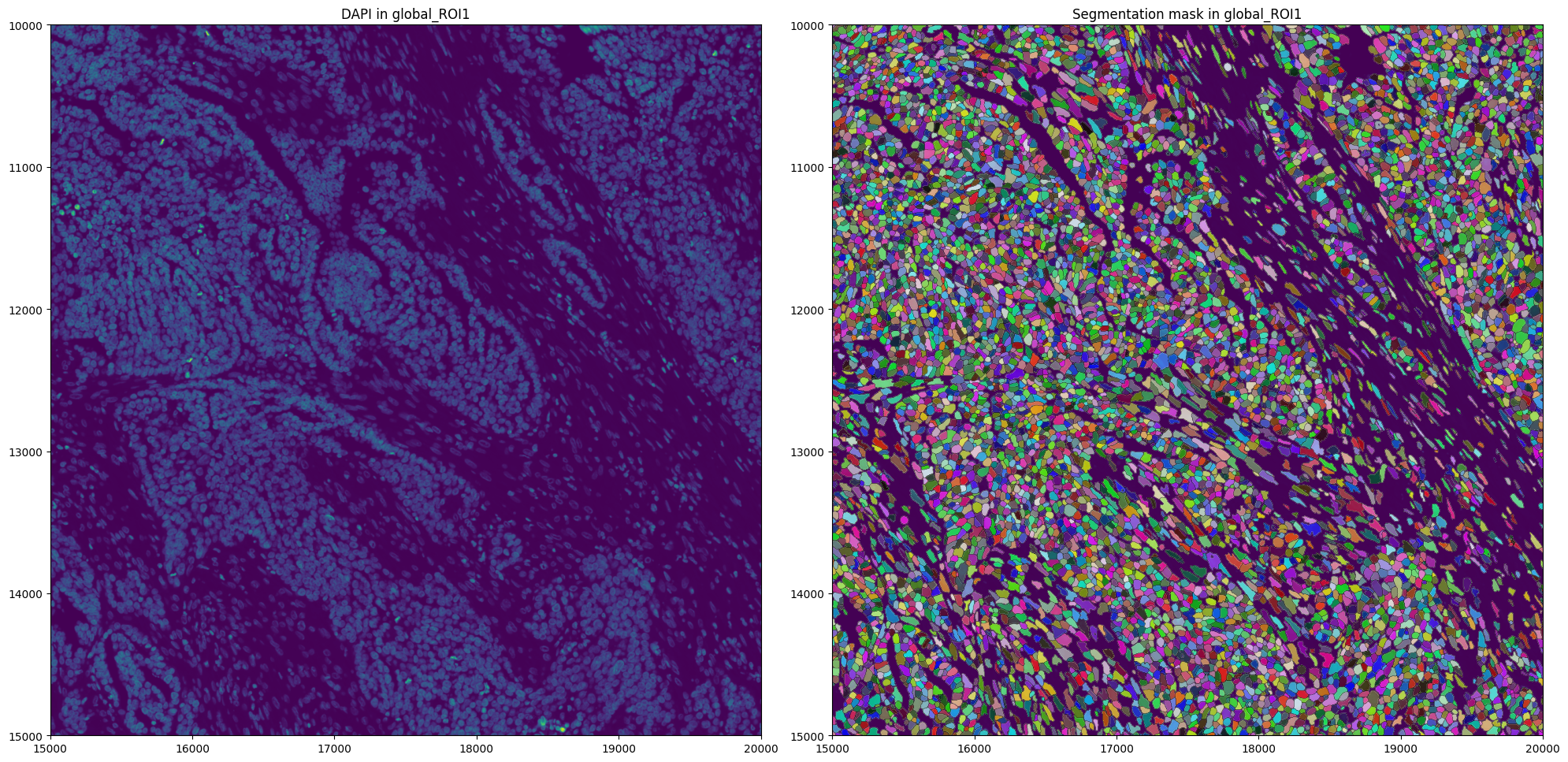
In many cases, you want to generate these segmentation masks yourself. Therefore, for illustration purposes, we show how to segment using Cellpose via harpy.im.segment, and how to generate an AnnData table using hp.tb.allocate.
Make sure to install Cellpose before running the following cell (pip install cellpose).
import os
import torch
from dask.distributed import Client, LocalCluster
if torch.cuda.is_available():
from dask_cuda import LocalCUDACluster # pip install dask-cuda
# One worker per GPU; each worker will be pinned to a single GPU.
cluster = LocalCUDACluster(
CUDA_VISIBLE_DEVICES=[0], # or [0,1],...etc
n_workers=1, # 2 if [0,1],...etc
threads_per_worker=1,
memory_limit="32GB",
local_directory=os.environ.get("TMPDIR"),
)
else:
# cpu/mps fall back
from dask.distributed import LocalCluster
cluster = LocalCluster(
n_workers=1
if torch.backends.mps.is_available()
else 8, # If mps/cuda device available, it is better to increase chunk size to maximal value that fits on the gpu, and set n_workers to 1.
# For this dummy example, we only have one chunk, so setting n_workers>1, has no effect.
threads_per_worker=1,
memory_limit="32GB",
local_directory=os.environ.get("TMPDIR"),
)
client = Client(cluster)
print(client.dashboard_link)
sdata = hp.im.segment(
sdata,
image_name="morphology_focus_global_ROI1", # note, currently cellpose only supports segmentation of 3 channels. So the channel at index -1 will not be used (AlphaSMA/Vimentin)
depth=200,
model=hp.im.cellpose_callable,
# parameters that will be passed to the callable cellpose_callable
diameter=50,
flow_threshold=0.9,
cellprob_threshold=-4,
output_labels_name="cell_labels_harpy_global_ROI1",
output_shapes_name=None,
crd=[15000, 20000, 10000, 15000],
# region to segment [x_min, xmax, y_min, y_max], must be defined in pixel coordinates, e.g. specify the pixel coordinate system
to_coordinate_system="global_ROI1",
overwrite=True,
)
client.close()
http://127.0.0.1:8787/status
Lets inspect the segmentation masks we generated.
import matplotlib.pyplot as plt
fig, axes = plt.subplots(1, 2, figsize=(20, 10))
channel = "DAPI"
image_name = "morphology_focus_global_ROI1"
labels_name = "cell_labels_harpy_global_ROI1"
crd = [15000, 20000, 10000, 15000]
to_coordinate_system = "global_ROI1"
render_images_kwargs = {"cmap": "viridis"}
render_labels_kwargs = {"fill_alpha": 0.6, "outline_alpha": 0.4}
show_kwargs = {"title": f"DAPI in {to_coordinate_system}", "colorbar": False}
hp.pl.plot_sdata(
sdata,
image_name=image_name,
crd=crd,
to_coordinate_system=to_coordinate_system,
channel=channel,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=axes[0],
)
# _ax.axis("off")
show_kwargs = {
"title": f"Segmentation mask (Harpy/Cellpose) in {to_coordinate_system}",
"colorbar": False,
}
hp.pl.plot_sdata(
sdata,
image_name=image_name,
labels_name=labels_name,
crd=crd,
to_coordinate_system=to_coordinate_system,
channel=channel,
render_images_kwargs=render_images_kwargs,
render_labels_kwargs=render_labels_kwargs,
show_kwargs=show_kwargs,
ax=axes[1],
)
# _ax.axis("off")
plt.tight_layout()
plt.show()
INFO Rasterizing image for faster rendering.
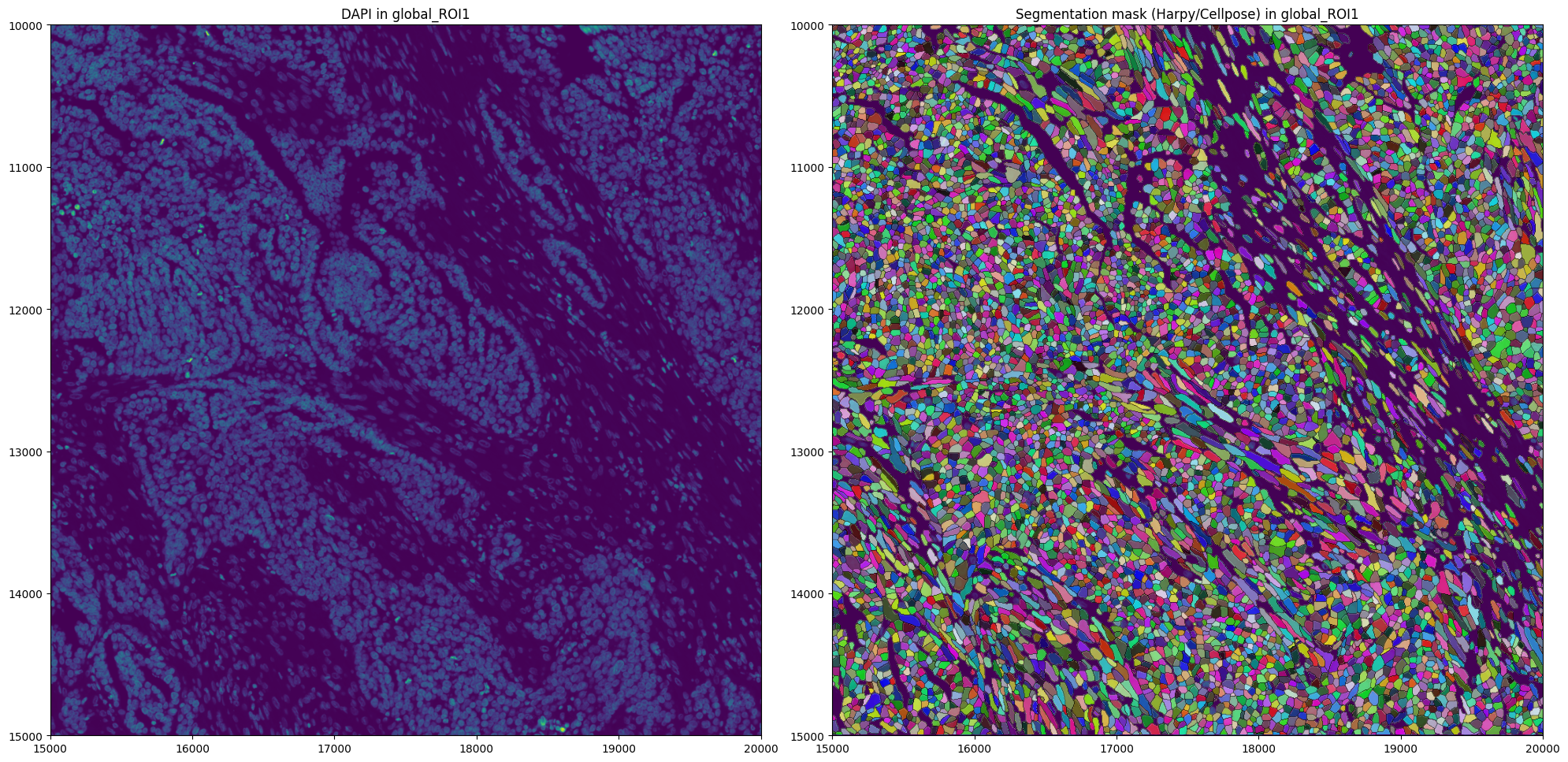
4. Create the AnnData table#
Below we illustrate how an AnnData table can be created using this custom generated segmentation mask via the harpy function harpy.tb.allocate.
from dask.distributed import Client, LocalCluster
cluster = LocalCluster(
n_workers=16,
threads_per_worker=1,
processes=True,
memory_limit="8GB",
local_directory=os.environ.get("TMPDIR"), # folder where dask will spill to disk
)
client = Client(cluster)
sdata = sdata = hp.tb.allocate(
sdata,
labels_name="cell_labels_harpy_global_ROI1",
points_name="transcripts_global_ROI1",
output_table_name="table_harpy_global_ROI1",
to_coordinate_system="global_ROI1", # Do allocation step in the intrinsic pixel coordinate system,
append=False,
overwrite=True,
update_shapes_elements=False,
)
client.close()
5. Preprocess the AnnData table#
We proceed working with the AnnData table provided by 10x Genomics. But the same logic applies to a custom generated AnnData table.
labels_name = "cell_labels_global_ROI1"
table_name = "table_global_ROI1"
points_name = "transcripts_global_ROI1"
filtered = hp.qc.analyse_genes_left_out(
sdata,
labels_name=labels_name,
table_name=table_name,
points_name=points_name,
to_coordinate_system=to_coordinate_system,
)
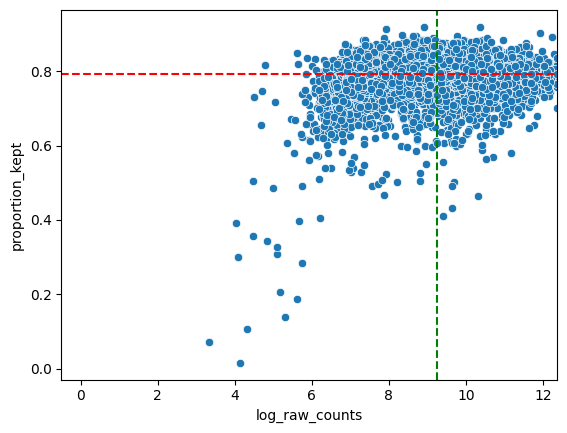
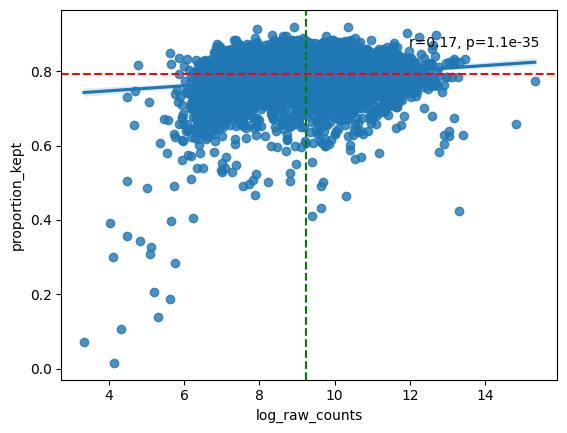
We proceed with filtering of cells with low transcript count, and genes with low cell count; normalization using cell size; a log transformation; and scaling such that the data has mean zero and a variance of one.
sdata = hp.tb.preprocess_transcriptomics(
sdata,
labels_name=labels_name,
table_name=table_name,
output_table_name="table_global_ROI1_preprocessed",
size_norm=True,
update_shapes_elements=False,
overwrite=True,
)
hp.pl.preprocess_transcriptomics(
sdata,
table_name="table_global_ROI1_preprocessed",
)
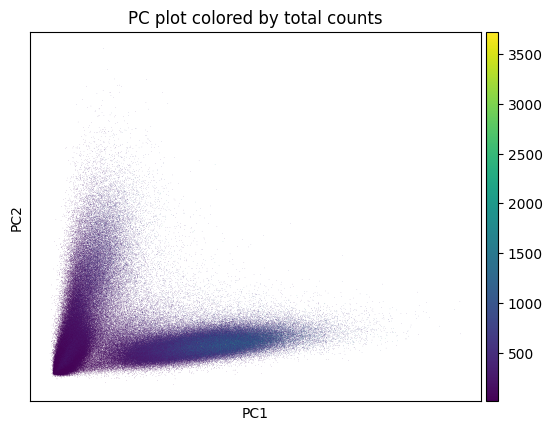
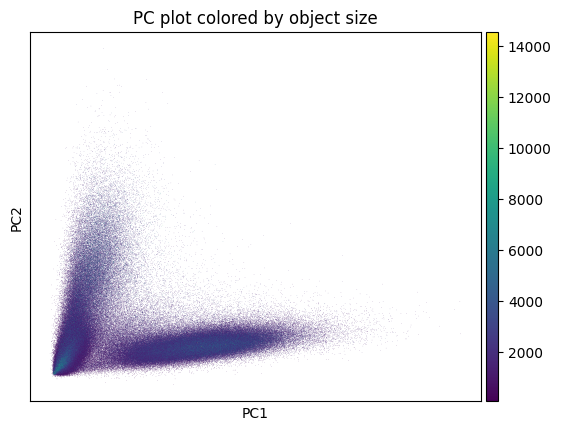
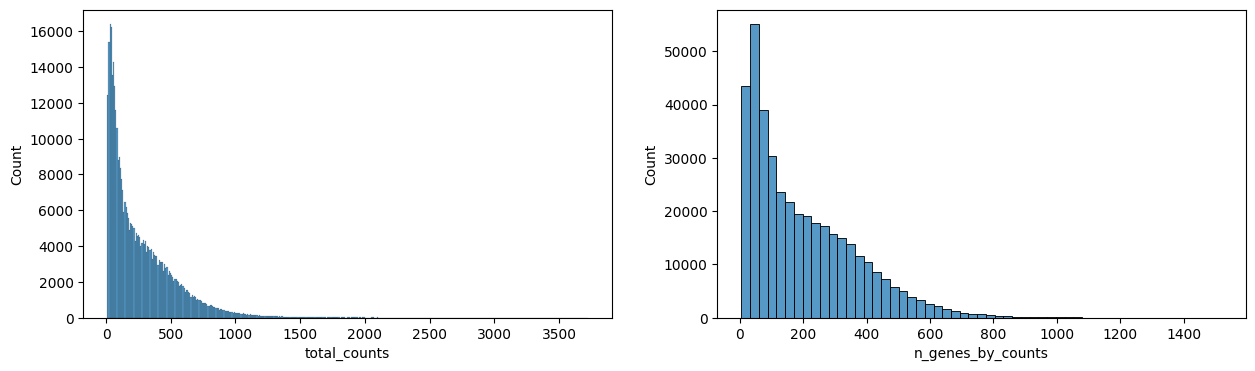
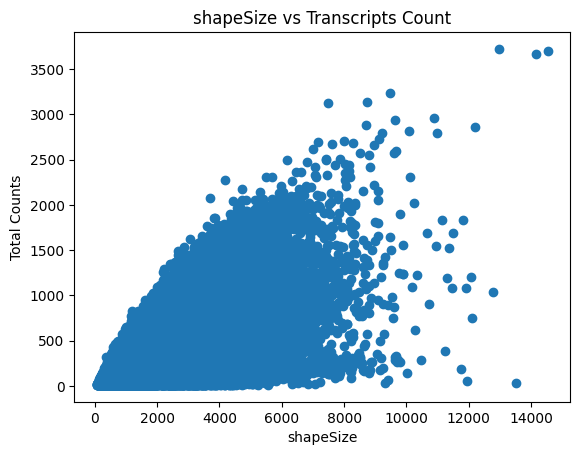
Visualize the expression after preprocessing.
# show expression after preprocessing;
import matplotlib.pyplot as plt
channel = "DAPI"
image_name = "morphology_focus_global_ROI1"
labels_name = "cell_labels_global_ROI1"
table_name = "table_global_ROI1_preprocessed"
fig, axes = plt.subplots(1, 1, figsize=(10, 10))
to_coordinate_system = to_coordinate_system
scale = None # auto pick scale by spatialdata-plot
color = "PLXNB1" # visualize expression of specific genes
crd = None
# if points in, query is slow, so remove them for plotting
sdata_to_plot = sdata.subset(element_names=[image_name, labels_name, table_name])
render_images_kwargs = {"cmap": "grey", "norm": None, "scale": scale}
render_labels_kwargs = {"fill_alpha": 0.5, "outline_alpha": 0.4, "scale": scale}
show_kwargs = {
"title": color,
"colorbar": False,
}
axes = hp.pl.plot_sdata(
sdata_to_plot,
image_name=image_name,
labels_name=labels_name,
table_name=table_name,
color=color,
channel=channel,
crd=crd,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=axes,
to_coordinate_system=to_coordinate_system,
)
plt.tight_layout()
plt.show()
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.

6. Leiden clustering.#
# takes around 40 minutes
# This can consume quite some RAM, as the AnnData table is in memory.
labels_name = "cell_labels_global_ROI1"
table_name = "table_global_ROI1_preprocessed"
sdata = hp.tb.leiden(
sdata,
labels_name=labels_name,
table_name=table_name,
output_table_name=table_name,
overwrite=True,
)
sdata.tables["table_global_ROI1_preprocessed"].obs.head()
| transcript_counts | control_probe_counts | genomic_control_counts | control_codeword_counts | unassigned_codeword_counts | deprecated_codeword_counts | total_counts | cell_area | nucleus_area | nucleus_count | ... | cell_ID | group | n_genes_by_counts | log1p_n_genes_by_counts | log1p_total_counts | pct_counts_in_top_2_genes | pct_counts_in_top_5_genes | n_counts | shapeSize | leiden | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cell_id | |||||||||||||||||||||
| aaaaebmm-1 | 303 | 0 | 0 | 0 | 0 | 31 | 303.0 | 44.659533 | 31.835157 | 1.0 | ... | 1 | Proliferative Tumor Cells | 246 | 5.509388 | 5.717028 | 7.260726 | 10.231023 | 303.0 | 989 | 3 |
| aaaafhpp-1 | 311 | 0 | 0 | 0 | 0 | 28 | 311.0 | 57.664533 | 38.247345 | 1.0 | ... | 2 | Tumor Cells | 255 | 5.545177 | 5.743003 | 2.893891 | 6.109325 | 311.0 | 1277 | 10 |
| aaaahcem-1 | 301 | 0 | 0 | 0 | 0 | 32 | 301.0 | 39.827814 | 27.590470 | 1.0 | ... | 3 | Proliferative Tumor Cells | 249 | 5.521461 | 5.710427 | 4.983389 | 8.637874 | 301.0 | 882 | 3 |
| aaaakeoi-1 | 369 | 0 | 0 | 0 | 0 | 58 | 369.0 | 60.870627 | 38.879533 | 1.0 | ... | 4 | Proliferative Tumor Cells | 306 | 5.726848 | 5.913503 | 3.252033 | 5.962060 | 369.0 | 1348 | 3 |
| aaaalald-1 | 295 | 0 | 0 | 0 | 0 | 47 | 295.0 | 50.484689 | 46.510939 | 1.0 | ... | 5 | Tumor Cells | 254 | 5.541264 | 5.690360 | 5.423729 | 8.813559 | 295.0 | 1118 | 10 |
5 rows × 23 columns
import scanpy as sc
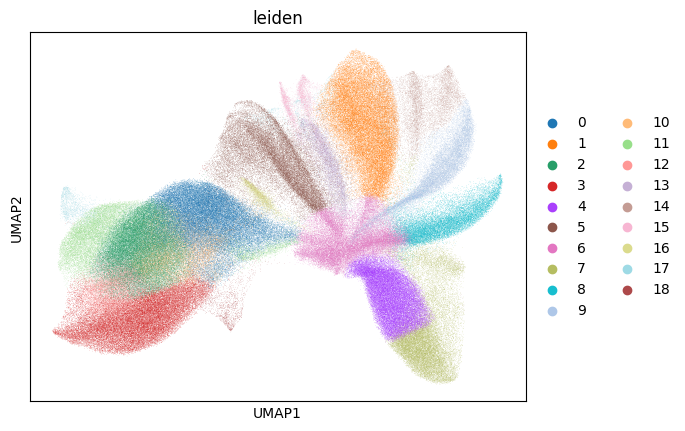
sc.pl.umap(sdata.tables["table_global_ROI1_preprocessed"], color=["leiden"], show=True)
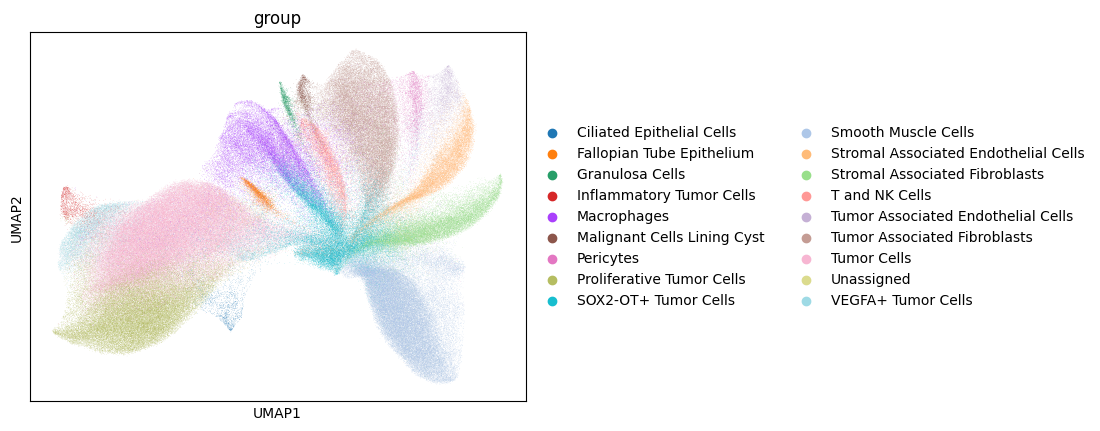
# this dataset is annotated, lets see how clusters correspond to cell type
sc.pl.umap(sdata.tables["table_global_ROI1_preprocessed"], color=["group"], show=True)
sc.pl.rank_genes_groups(
sdata.tables["table_global_ROI1_preprocessed"], n_genes=8, sharey=False, show=False
)
[<Axes: title={'center': '0 vs. rest'}, ylabel='score'>,
<Axes: title={'center': '1 vs. rest'}>,
<Axes: title={'center': '2 vs. rest'}>,
<Axes: title={'center': '3 vs. rest'}>,
<Axes: title={'center': '4 vs. rest'}, ylabel='score'>,
<Axes: title={'center': '5 vs. rest'}>,
<Axes: title={'center': '6 vs. rest'}>,
<Axes: title={'center': '7 vs. rest'}>,
<Axes: title={'center': '8 vs. rest'}, ylabel='score'>,
<Axes: title={'center': '9 vs. rest'}>,
<Axes: title={'center': '10 vs. rest'}>,
<Axes: title={'center': '11 vs. rest'}>,
<Axes: title={'center': '12 vs. rest'}, ylabel='score'>,
<Axes: title={'center': '13 vs. rest'}>,
<Axes: title={'center': '14 vs. rest'}>,
<Axes: title={'center': '15 vs. rest'}>,
<Axes: title={'center': '16 vs. rest'}, xlabel='ranking', ylabel='score'>,
<Axes: title={'center': '17 vs. rest'}, xlabel='ranking'>,
<Axes: title={'center': '18 vs. rest'}, xlabel='ranking'>]

import matplotlib.pyplot as plt
channel = "DAPI"
image_name = "morphology_focus_global_ROI1"
labels_name = "cell_labels_global_ROI1"
table_name = "table_global_ROI1_preprocessed"
to_coordinate_system = "global_ROI1"
to_coordinate_system = to_coordinate_system
scale = None # auto pick scale by spatialdata-plot
crd = None
color = "leiden"
fig, ax = plt.subplots(1, 1, figsize=(10, 10))
# if points in, query is slow, so remove them for plotting
sdata_to_plot = sdata.subset(element_names=[image_name, labels_name, table_name])
render_images_kwargs = {"cmap": "grey", "norm": None, "scale": scale}
render_labels_kwargs = {"fill_alpha": 0.5, "outline_alpha": 0.4, "scale": scale}
show_kwargs = {
"title": color,
"colorbar": False,
}
ax = hp.pl.plot_sdata(
sdata_to_plot,
image_name=image_name,
labels_name=labels_name,
table_name=table_name,
color=color,
channel=channel,
crd=crd,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=ax,
to_coordinate_system=to_coordinate_system,
)
# _ax.axis("off")
plt.tight_layout()
plt.show()
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.

import matplotlib.pyplot as plt
channel = "DAPI"
image_name = "morphology_focus_global_ROI1"
labels_name = "cell_labels_global_ROI1"
table_name = "table_global_ROI1_preprocessed"
to_coordinate_system = "global_ROI1"
to_coordinate_system = to_coordinate_system
scale = None # auto pick scale by spatialdata-plot
crd = None
color = "group"
fig, ax = plt.subplots(1, 1, figsize=(20, 20))
# if points in, query is slow, so remove them for plotting
sdata_to_plot = sdata.subset(element_names=[image_name, labels_name, table_name])
render_images_kwargs = {"cmap": "grey", "norm": None, "scale": scale}
render_labels_kwargs = {"fill_alpha": 0.5, "outline_alpha": 0.4, "scale": scale}
show_kwargs = {
"title": color,
"colorbar": False,
}
ax = hp.pl.plot_sdata(
sdata_to_plot,
image_name=image_name,
labels_name=labels_name,
table_name=table_name,
color=color,
channel=channel,
crd=crd,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=ax,
to_coordinate_system=to_coordinate_system,
)
# _ax.axis("off")
plt.tight_layout()
plt.show()
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.

Note the correspondence between Leiden cluster 17, and the Inflammatory Tumor Cells.
We also estimate the correlation between leiden clusters and the annotated cell type.
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import seaborn as sns
from scipy.stats import chi2_contingency
table_name = "table_global_ROI1_preprocessed"
cluster_key = "leiden"
cell_type_key = "group"
df = sdata[table_name].obs
df = df[~df[cell_type_key].isna()]
contingency = pd.crosstab(df[cell_type_key], df[cluster_key])
# chi squared test
chi2, p, dof, expected = chi2_contingency(contingency)
# cramers v
def _cramers_v(confusion_matrix):
chi2 = chi2_contingency(confusion_matrix)[0]
n = confusion_matrix.sum().sum()
phi2 = chi2 / n
r, k = confusion_matrix.shape
return np.sqrt(phi2 / min(k - 1, r - 1))
print("Cramers v:", _cramers_v(contingency))
print("Chi-squared statistic:", chi2)
print("p-value:", p)
plt.figure(figsize=(12, 6))
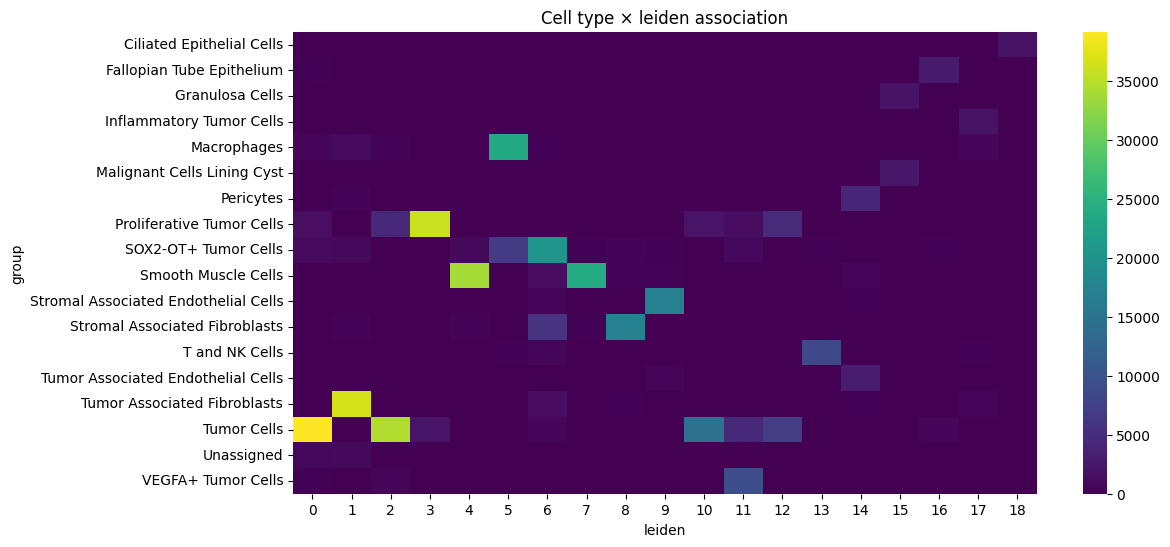
sns.heatmap(contingency, cmap="viridis")
plt.title(f"Cell type × {cluster_key} association")
plt.show()
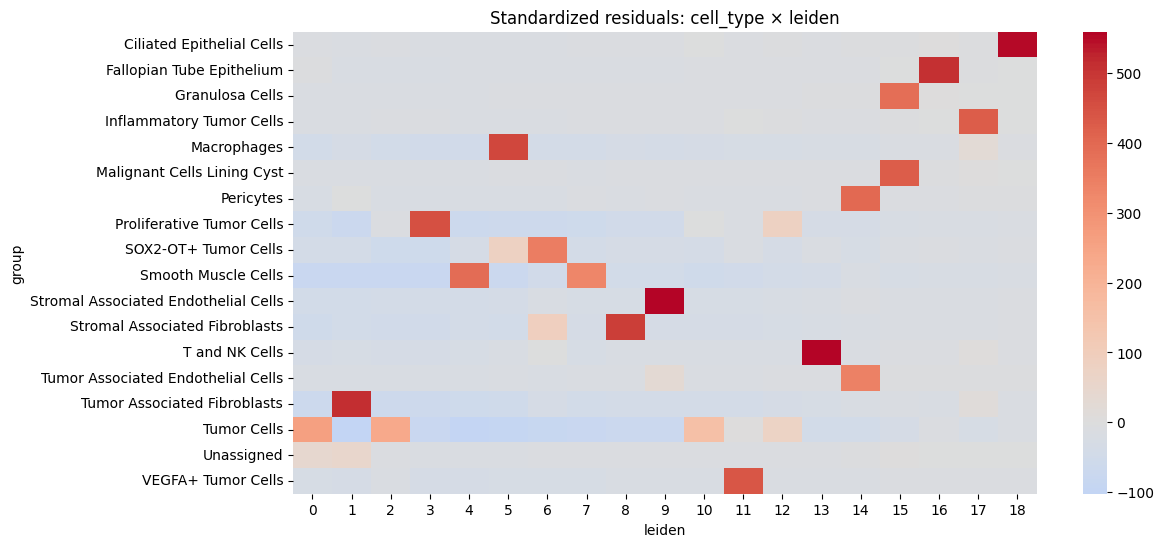
# Calculate standardized residuals
residuals = (contingency - expected) / np.sqrt(expected)
residuals_df = pd.DataFrame(
residuals, index=contingency.index, columns=contingency.columns
)
resid_long = residuals_df.stack().reset_index()
resid_long.columns = ["cell_type", cluster_key, "std_residual"]
# Sort by strength of deviation
resid_long["abs_resid"] = resid_long["std_residual"].abs()
top_effects = resid_long.sort_values("abs_resid", ascending=False).head(20)
print(top_effects)
plt.figure(figsize=(12, 6))
sns.heatmap(residuals_df, center=0, cmap="coolwarm", annot=False)
plt.title(f"Standardized residuals: cell_type × {cluster_key}")
plt.show()
Cramers v: 0.7613470480652001
Chi-squared statistic: 3925208.4534459906
p-value: 0.0
cell_type leiden std_residual abs_resid
199 Stromal Associated Endothelial Cells 9 558.842983 558.842983
241 T and NK Cells 13 554.391652 554.391652
18 Ciliated Epithelial Cells 18 553.300254 553.300254
267 Tumor Associated Fibroblasts 1 513.570314 513.570314
35 Fallopian Tube Epithelium 16 505.783565 505.783565
217 Stromal Associated Fibroblasts 8 485.604699 485.604699
81 Macrophages 5 466.487412 466.487412
136 Proliferative Tumor Cells 3 449.341736 449.341736
334 VEGFA+ Tumor Cells 11 438.169924 438.169924
110 Malignant Cells Lining Cyst 15 425.370919 425.370919
74 Inflammatory Tumor Cells 17 425.115462 425.115462
128 Pericytes 14 400.937651 400.937651
175 Smooth Muscle Cells 4 389.203349 389.203349
53 Granulosa Cells 15 385.049195 385.049195
158 SOX2-OT+ Tumor Cells 6 351.807910 351.807910
261 Tumor Associated Endothelial Cells 14 343.997358 343.997358
178 Smooth Muscle Cells 7 327.645762 327.645762
285 Tumor Cells 0 259.965609 259.965609
287 Tumor Cells 2 235.059284 235.059284
295 Tumor Cells 10 154.009542 154.009542