Highly-multiplexed image analysis with Harpy and Kronos.#
This notebook reimplements parts of the code from the KRONOS repository to enable interoperability with SpatialData. Feature extraction using the KRONOS model has been optimized with Dask for efficient scaling to gigapixel-scale images. We also refer to the KRONOS paper by Shaban M. et al (2025).
We will use an annotated CODEX datataset (Shaban M. et al. (2023)).
To obtain a mapping between your data specific marker names and the marker names used by the KRONOS foundation model, we refer to this notebook.
To run this notebook, make sure to install harpy and kronos.
uv venv .venv_harpy_kronos --python=3.12
source .venv_harpy_kronos/bin/activate
uv pip install "git+https://github.com/saeyslab/harpy.git@main"
uv pip install "git+https://github.com/mahmoodlab/KRONOS.git#egg=kronos[tutorials]"
import os
import harpy as hp
# This will download approximately 24GB. By setting the environment varialble 'HARPY_POOCH_CACHE' in a .env file, one can specify where this data will be cached.
sdata = hp.datasets.codex_example()
path = os.environ.get("SCRATCHDIR") # choose an output directory of choice.
sdata.write(os.path.join(path, "sdata_codex.zarr"))
# Fetch the csv file containing the mapping between the data specific marker names and the marker names used by the Kronos model.
registry = hp.datasets.get_registry()
matched_channels_path = registry.fetch("proteomics/codex/chl_maps_dataset/marker_metadata_mapped.csv")
Before running the Harpy function hp.tb.featurize, please make sure the image and the segmentation mask, have the same spatial chunksize on disk.
Using a smaller chunksize in c, y and x will reduce RAM usage.
If not, you could rechunk as follows:
sdata = hp.im.add_image(
sdata,
arr=sdata["image"][ "scale0" ]["image"].data.rechunk( ( 10,2048,2048 ) ),
output_image_name = "image",
scale_factors=[2,2,2,2],
c_coords = sdata["image"][ "scale0" ]["image"].c.data,
overwrite=True,
)
sdata = hp.im.add_labels(
sdata,
arr=sdata["segmentation_mask"][ "scale0" ]["image"].data.rechunk( ( 2048,2048 ) ),
output_labels_name = "segmentation_mask",
scale_factors=[2,2,2,2],
overwrite=True,
)
display(sdata["image"]["scale0"]["image"])
display(sdata["segmentation_mask"]["scale0"]["image"])
<xarray.DataArray 'image' (c: 49, y: 8011, x: 8085)> Size: 6GB
dask.array<from-zarr, shape=(49, 8011, 8085), dtype=uint16, chunksize=(10, 2048, 2048), chunktype=numpy.ndarray>
Coordinates:
* c (c) <U11 2kB 'BCL-2' 'CCR6' 'CD11B' ... 'A-SMA' 'B-CATENIN'
* y (y) float64 64kB 0.5 1.5 2.5 3.5 ... 8.008e+03 8.01e+03 8.01e+03
* x (x) float64 65kB 0.5 1.5 2.5 3.5 ... 8.082e+03 8.084e+03 8.084e+03
Attributes:
transform: {'global': Identity }<xarray.DataArray 'image' (y: 8011, x: 8085)> Size: 259MB
dask.array<from-zarr, shape=(8011, 8085), dtype=int32, chunksize=(2048, 2048), chunktype=numpy.ndarray>
Coordinates:
* y (y) float64 64kB 0.5 1.5 2.5 3.5 ... 8.008e+03 8.01e+03 8.01e+03
* x (x) float64 65kB 0.5 1.5 2.5 3.5 ... 8.082e+03 8.084e+03 8.084e+03
Attributes:
transform: {'global': Identity }The AnnData table "table_intensities"in the SpatialData object sdata contains the main intensity (in sdata["table_intensities"].X) for each instance in the segmentation mask segmentation_mask. These mean intensities were calculated as follows:
hp.tb.allocate_intensity(
sdata,
image_name="image",
labels_name="segmentation_mask",
output_table_name=table_name,
mode="mean",
calculate_center_of_mass=True,
overwrite=True,
)
sdata["table_intensities"].to_df().head()
| channels | BCL-2 | CCR6 | CD11B | CD11C | CD15 | CD16 | CD162 | CD163 | CD2 | CD20 | ... | PD-L1 | PODOPLANIN | T-BET | TCR-G-D | TCRB | TIM-3 | VISA | VIMENTIN | A-SMA | B-CATENIN |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cells | |||||||||||||||||||||
| 1_segmentation_mask_3314222a | 34272.214844 | 1579.261963 | 1.000000 | 1.000000 | 1.000000 | 1088.190430 | 37.214287 | 113.309525 | 7561.904785 | 51194.882812 | ... | 630.404785 | 591.785706 | 1664.023804 | 5099.714355 | 531.071411 | 60.666668 | 1.000000 | 2658.666748 | 4.547619 | 58.119049 |
| 2_segmentation_mask_3314222a | 0.000000 | 325.746033 | 2291.952393 | 14611.238281 | 1304.349243 | 41.190475 | 823.365051 | 151.888885 | 33.047619 | 0.412698 | ... | 1127.000000 | 1.000000 | 118.333336 | 1740.126953 | 194.634918 | 1025.888916 | 402.714294 | 16652.603516 | 2.539683 | 268.000000 |
| 3_segmentation_mask_3314222a | 2622.344727 | 344.517242 | 2660.862061 | 2214.655273 | 3181.965576 | 1082.896606 | 1.000000 | 113.724136 | 88.724136 | 1043.000000 | ... | 1177.758667 | 1.000000 | 273.344818 | 1515.206909 | 0.000000 | 45.965519 | 1001.137939 | 1358.206909 | 6.000000 | 105.206894 |
| 4_segmentation_mask_3314222a | 0.250000 | 32.906250 | 2395.468750 | 4671.671875 | 0.453125 | 705.953125 | 80.031250 | 3040.734375 | 1629.125000 | 1.000000 | ... | 364.453125 | 31.156250 | 30.375000 | 0.000000 | 174.687500 | 1361.390625 | 503.000000 | 1070.484375 | 182.000000 | 283.906250 |
| 5_segmentation_mask_3314222a | 8693.557617 | 282.901642 | 1.000000 | 210.622955 | 1.000000 | 41.606556 | 3547.672119 | 168.672134 | 6995.393555 | 2731.491699 | ... | 186.098358 | 60.786884 | 882.737732 | 1170.868896 | 1991.655762 | 122.836067 | 0.983607 | 4394.360840 | 30.295082 | 0.000000 |
5 rows × 49 columns
import pandas as pd
matched_channels = pd.read_csv(matched_channels_path)
matched_channels.head()
| marker_name | marker_name_pretrained | marker_id_pretrained | marker_mean | marker_std | marker_id | |
|---|---|---|---|---|---|---|
| 0 | BCL-2 | BCL2 | 150 | 0.047104 | 0.060276 | 0 |
| 1 | CCR6 | CCR6 | 166 | 0.044867 | 0.042833 | 1 |
| 2 | CD11B | CD11B | 180 | 0.032169 | 0.052366 | 2 |
| 3 | CD11C | CD11C | 182 | 0.019039 | 0.044336 | 3 |
| 4 | CD15 | CD15 | 194 | 0.016322 | 0.040416 | 4 |
from dask.distributed import Client, LocalCluster
cluster = LocalCluster(
n_workers=4,
threads_per_worker=1,
memory_limit="32GB",
local_directory=os.environ.get("SCRATCHDIR"), # directory where dask will spill to disk
)
client = Client(cluster)
print(client.dashboard_link)
http://127.0.0.1:8787/status
import pandas as pd
import harpy as hp
# Hugging Face authentication token needs to be provided when downloading the model from the Hugging Face hub.
HF_AUTH_TOKEN = os.environ.get("HF_AUTH_TOKEN")
# you could choose to specify a local path as the checkpoint path for the kronos model. By default it will download the kronos model from the Hugging Face hub "hf_hub:MahmoodLab/kronos".
# checkpoint_path = "/data/groups/technologies/spatial.catalyst/Arne/kronos_example_data/models--MahmoodLab--kronos/snapshots/8edc2719ad67b2e2b766073b35c6cf8e6f5da516/kronos_vits16_model.pt"
# you could choose to set markers_to_keep = None, then all markers will be used.
markers_to_keep = [
"DAPI-01",
"CD11B",
"CD11C",
"CD15",
"CD163",
"CD20",
"CD206",
"CD30",
"CD31",
"CD4",
"CD56",
"CD68",
"CD7",
"CD8",
"CYTOKERITIN",
"FOXP3",
"MCT",
"PODOPLANIN",
]
matched_channels = matched_channels[matched_channels["marker_name"].isin(markers_to_keep)]
# keyword arguments passed to hp.utils.kronos_embedding
model_kwargs = {
"hf_auth_token": HF_AUTH_TOKEN,
"matched_channels": matched_channels,
"max_value": 65535.0,
"do_instance_embedding": False,
}
table_name = "table_intensities"
sdata = hp.tb.featurize(
sdata,
image_name="image",
labels_name="segmentation_mask",
table_name=table_name,
output_table_name=table_name,
depth=200,
diameter=16 * 4, # kronos patch sizes must be multiples of 16
embedding_dimension=384
* matched_channels.shape[0], # all channels are matched, so it is embedding_dim*num_channels
model=hp.utils.kronos_embedding,
model_kwargs=model_kwargs,
batch_size=250,
overwrite=True,
)
client.close()
sdata["table_intensities"].obsm["embedding"].shape # -> kronos embedding
(152815, 6912)
from sklearn.cluster import KMeans
kmeans = KMeans(
n_clusters=20,
random_state=0,
)
labels = kmeans.fit_predict(sdata[table_name].obsm["embedding"])
sdata[table_name].obs["kronos"] = labels
sdata[table_name].obs["kronos"] = sdata[table_name].obs["kronos"].astype("category")
# back cluster ids to zarr
sdata = hp.tb.add_table(
sdata,
adata=sdata[table_name],
output_table_name=table_name,
region=["segmentation_mask"],
overwrite=True,
)
# normalize before calculating leiden clusters
sdata = hp.tb.preprocess_proteomics(
sdata,
labels_name="segmentation_mask",
table_name=table_name,
output_table_name=f"{table_name}_normalized",
size_norm=False,
log1p=False,
q=0.99,
max_value_q=1,
overwrite=True,
)
sdata[f"{table_name}_normalized"].to_df().head()
| channels | BCL-2 | CCR6 | CD11B | CD11C | CD15 | CD16 | CD162 | CD163 | CD2 | CD20 | ... | PD-L1 | PODOPLANIN | T-BET | TCR-G-D | TCRB | TIM-3 | VISA | VIMENTIN | A-SMA | B-CATENIN |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cells | |||||||||||||||||||||
| 1_segmentation_mask_3314222a | 79.668221 | 62.177567 | 0.011111 | 0.004568 | 0.003408 | 14.284677 | 0.796612 | 1.115642 | 36.489056 | 100.000000 | ... | 8.457295 | 11.425946 | 24.895376 | 68.573784 | 16.188705 | 0.666807 | 0.045563 | 13.841380 | 4.506205 | 5.740132 |
| 2_segmentation_mask_3314222a | 0.000000 | 12.825038 | 25.464931 | 66.745941 | 4.445347 | 0.540707 | 17.625029 | 1.495494 | 0.159467 | 0.000900 | ... | 15.119448 | 0.019308 | 1.770379 | 23.398777 | 5.933077 | 11.275876 | 18.348717 | 86.695709 | 2.516554 | 26.469042 |
| 3_segmentation_mask_3314222a | 6.095829 | 13.564084 | 29.563736 | 10.116818 | 10.844443 | 14.215184 | 0.021406 | 1.119725 | 0.428128 | 2.274806 | ... | 15.800408 | 0.019308 | 4.089499 | 20.374371 | 0.000000 | 0.505222 | 45.614468 | 7.071009 | 5.945360 | 10.390767 |
| 4_segmentation_mask_3314222a | 0.000581 | 1.295561 | 26.615061 | 21.340773 | 0.001544 | 9.267047 | 1.713156 | 29.938982 | 7.861145 | 0.002181 | ... | 4.889379 | 0.601552 | 0.454439 | 0.000000 | 5.325017 | 14.963484 | 22.917997 | 5.573087 | 100.000000 | 28.040024 |
| 5_segmentation_mask_3314222a | 20.208799 | 11.138199 | 0.011111 | 0.962152 | 0.003408 | 0.546169 | 75.941795 | 1.660741 | 33.755424 | 5.957442 | ... | 2.496632 | 1.173647 | 13.206595 | 15.744198 | 60.711853 | 1.350131 | 0.044816 | 22.877638 | 30.019194 | 0.000000 |
5 rows × 49 columns
# takes approx 20 minutes
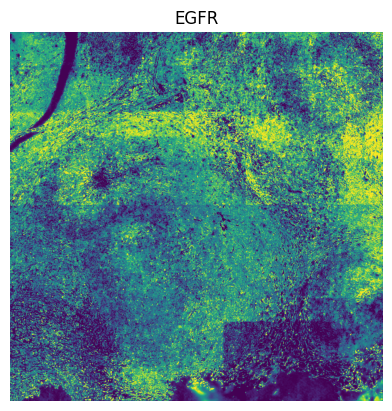
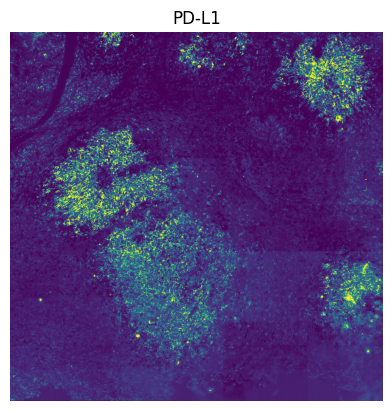
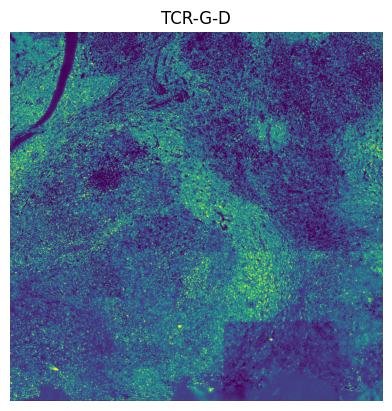
# We do leiden clustering with these channels filtered out: [ "EGFR", "FOXP3", "PD-L1", "T-BET", "TCR-G-D", "CD56", "CD69" ], because there are tiling effects in them.
subset_channels = [
_channel
for _channel in sdata[f"{table_name}_normalized"].var_names
if _channel not in ["EGFR", "FOXP3", "PD-L1", "T-BET", "TCR-G-D", "CD56", "CD69"]
]
sdata = hp.tb.leiden(
sdata,
labels_name="segmentation_mask",
table_name=f"{table_name}_normalized",
output_table_name=f"{table_name}_normalized_leiden", # write to new slot, because we subset the channels
index_names_var=subset_channels,
key_added="leiden",
overwrite=True,
)
sdata[f"{table_name}_normalized"].to_df().head()
| channels | BCL-2 | CCR6 | CD11B | CD11C | CD15 | CD16 | CD162 | CD163 | CD2 | CD20 | ... | PD-L1 | PODOPLANIN | T-BET | TCR-G-D | TCRB | TIM-3 | VISA | VIMENTIN | A-SMA | B-CATENIN |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cells | |||||||||||||||||||||
| 1_segmentation_mask_3314222a | 79.668221 | 62.177567 | 0.011111 | 0.004568 | 0.003408 | 14.284677 | 0.796612 | 1.115642 | 36.489056 | 100.000000 | ... | 8.457295 | 11.425946 | 24.895376 | 68.573784 | 16.188705 | 0.666807 | 0.045563 | 13.841380 | 4.506205 | 5.740132 |
| 2_segmentation_mask_3314222a | 0.000000 | 12.825038 | 25.464931 | 66.745941 | 4.445347 | 0.540707 | 17.625029 | 1.495494 | 0.159467 | 0.000900 | ... | 15.119448 | 0.019308 | 1.770379 | 23.398777 | 5.933077 | 11.275876 | 18.348717 | 86.695709 | 2.516554 | 26.469042 |
| 3_segmentation_mask_3314222a | 6.095829 | 13.564084 | 29.563736 | 10.116818 | 10.844443 | 14.215184 | 0.021406 | 1.119725 | 0.428128 | 2.274806 | ... | 15.800408 | 0.019308 | 4.089499 | 20.374371 | 0.000000 | 0.505222 | 45.614468 | 7.071009 | 5.945360 | 10.390767 |
| 4_segmentation_mask_3314222a | 0.000581 | 1.295561 | 26.615061 | 21.340773 | 0.001544 | 9.267047 | 1.713156 | 29.938982 | 7.861145 | 0.002181 | ... | 4.889379 | 0.601552 | 0.454439 | 0.000000 | 5.325017 | 14.963484 | 22.917997 | 5.573087 | 100.000000 | 28.040024 |
| 5_segmentation_mask_3314222a | 20.208799 | 11.138199 | 0.011111 | 0.962152 | 0.003408 | 0.546169 | 75.941795 | 1.660741 | 33.755424 | 5.957442 | ... | 2.496632 | 1.173647 | 13.206595 | 15.744198 | 60.711853 | 1.350131 | 0.044816 | 22.877638 | 30.019194 | 0.000000 |
5 rows × 49 columns
table_name
'table_intensities'
Add the calculated leiden cluster ids to the table element where we did not subset the channels. This allows us to calculate mean intensity on all channels layer on.
import pandas as pd
from spatialdata.models import TableModel
instance_key = sdata[table_name].uns[TableModel.ATTRS_KEY][TableModel.INSTANCE_KEY]
region_key = sdata[table_name].uns[TableModel.ATTRS_KEY][TableModel.REGION_KEY_KEY]
index = sdata[f"{table_name}_normalized"].obs.index
name = sdata[f"{table_name}_normalized"].obs.index.name
if "leiden" in sdata[f"{table_name}_normalized"].obs.columns:
sdata[f"{table_name}_normalized"].obs.drop(columns=["leiden"], inplace=True)
sdata[f"{table_name}_normalized"].obs = pd.merge(
sdata[f"{table_name}_normalized"].obs,
sdata[f"{table_name}_normalized_leiden"].obs[["leiden", region_key, instance_key]],
on=[instance_key, region_key],
how="inner",
)
sdata[f"{table_name}_normalized"].obs.index = index
sdata[f"{table_name}_normalized"].obs.index.name = name
# back to zarr store
sdata = hp.tb.add_table(
sdata,
adata=sdata[f"{table_name}_normalized"],
output_table_name=f"{table_name}_normalized",
region=["segmentation_mask"],
overwrite=True,
)
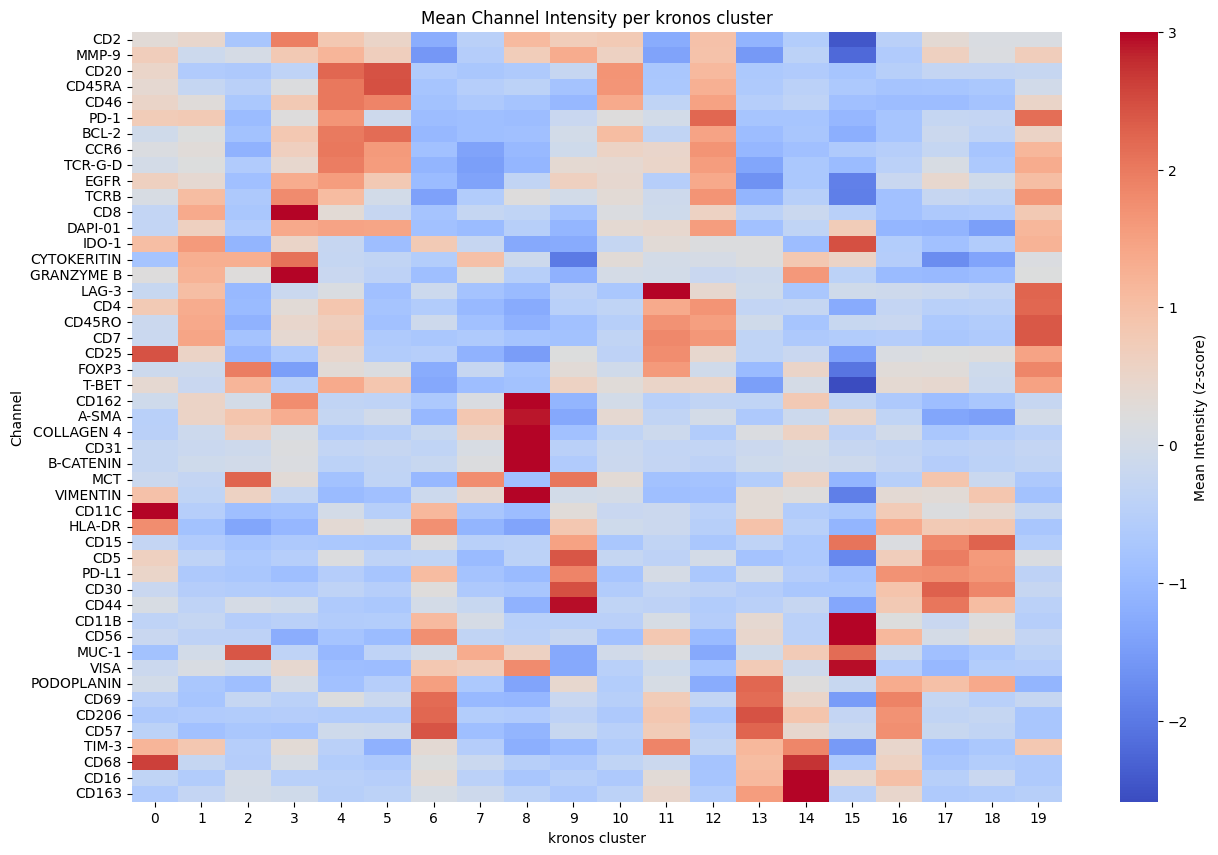
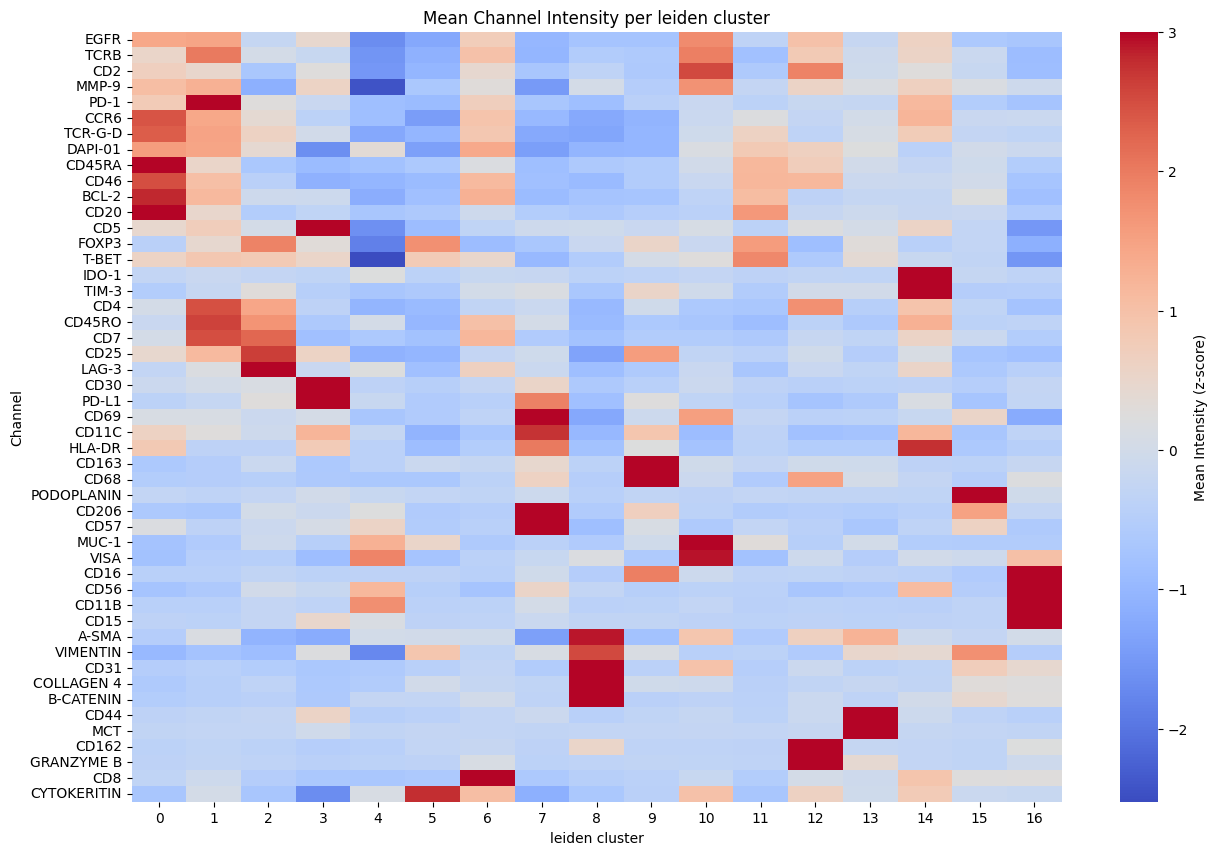
for _cluster in ["kronos", "leiden"]:
sdata = hp.tb.cluster_intensity(
sdata,
table_name=f"{table_name}_normalized",
labels_name="segmentation_mask",
output_table_name=f"{table_name}_normalized",
cluster_key=_cluster,
layer_mean_intensities="raw_counts", # layer in adata.layers that holds the mean intensities
overwrite=True,
)
hp.pl.cluster_intensity_heatmap(
sdata,
table_name=f"{table_name}_normalized",
cluster_key=_cluster,
figsize=(15, 10),
)
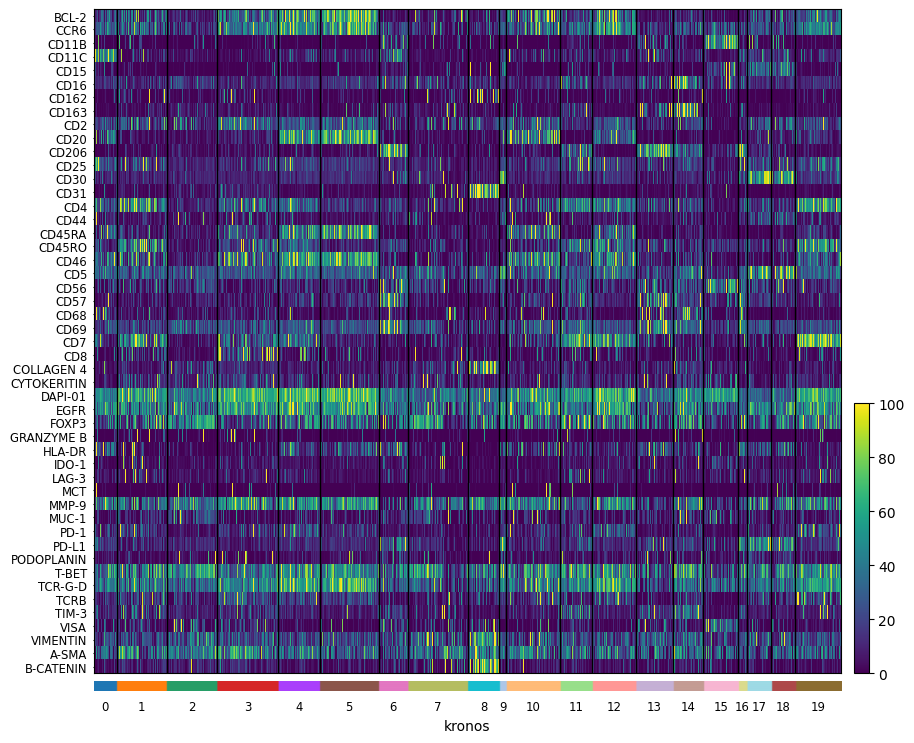
import scanpy as sc
from matplotlib.colors import Normalize
norm = Normalize(vmax=100, clip=False)
sc.pl.heatmap(
sdata[f"{table_name}_normalized"],
var_names=sdata[f"{table_name}_normalized"].var_names,
groupby="kronos",
swap_axes=True,
norm=norm,
use_raw=False,
)
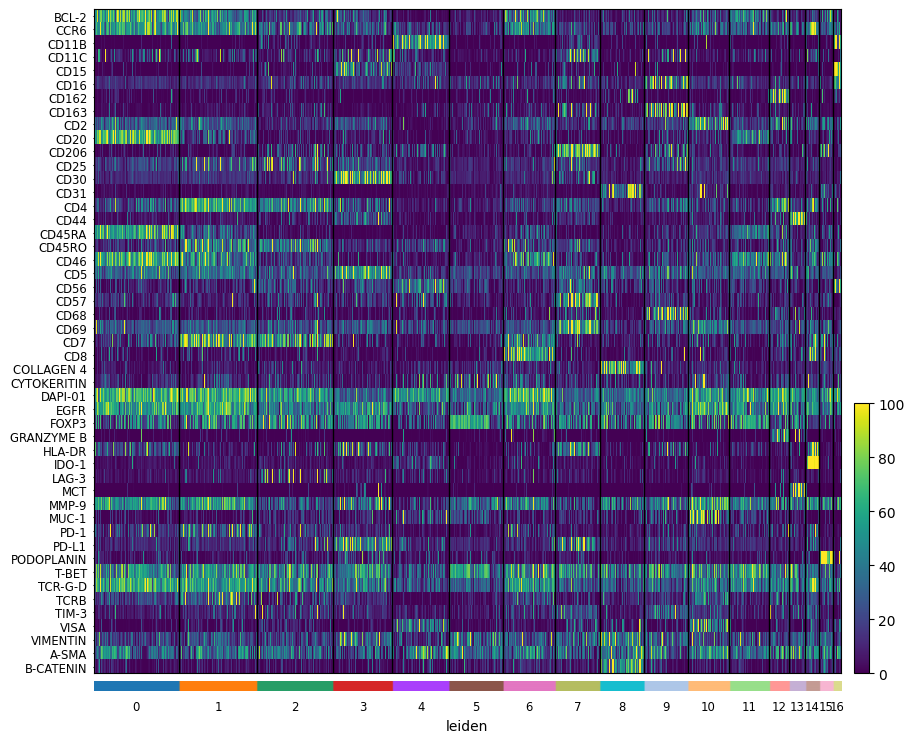
sc.pl.heatmap(
sdata[f"{table_name}_normalized"],
var_names=sdata[f"{table_name}_normalized"].var_names,
groupby="leiden",
swap_axes=True,
norm=norm,
use_raw=False,
)
sdata[f"{table_name}_normalized"].obsm["X_umap"] = sdata[f"{table_name}_normalized_leiden"].obsm["X_umap"]
import scanpy as sc
from matplotlib.colors import Normalize
sc.pl.umap(sdata[f"{table_name}_normalized"], color=["cell_type"])
for _cluster_key in ["kronos", "leiden"]:
sc.pl.umap(sdata[f"{table_name}_normalized"], color=[_cluster_key])
norm = Normalize(vmax=100, clip=False)
sc.pl.umap(
sdata[f"{table_name}_normalized"], color=["CD30", "PD-L1", "CD5"], norm=norm, use_raw=False
) # tumor markers
sc.pl.umap(sdata[f"{table_name}_normalized"], color=["CD11B", "CD11C", "CD15"], norm=norm, use_raw=False)
# note that these umaps are biased towards the leiden clusters (clusters will look more spatially seperated), because umaps are calculated on the same knn graph as the leiden clusters.
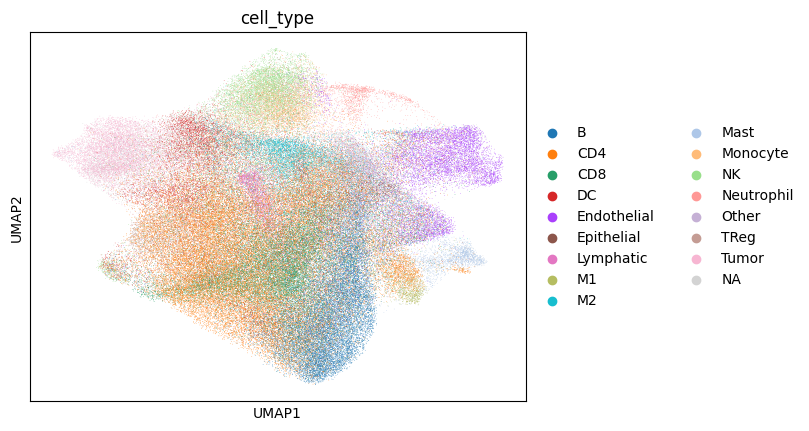
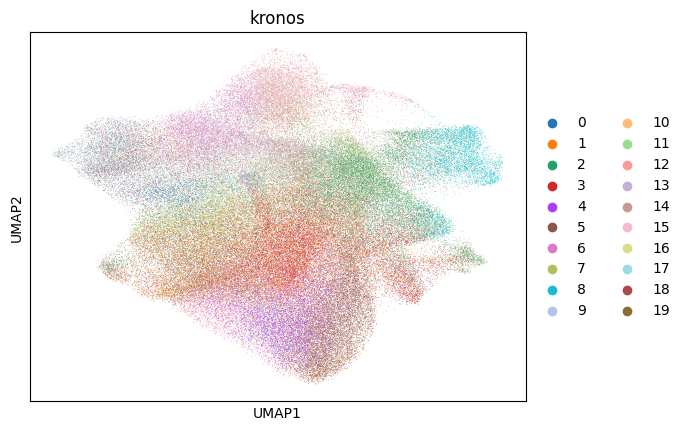
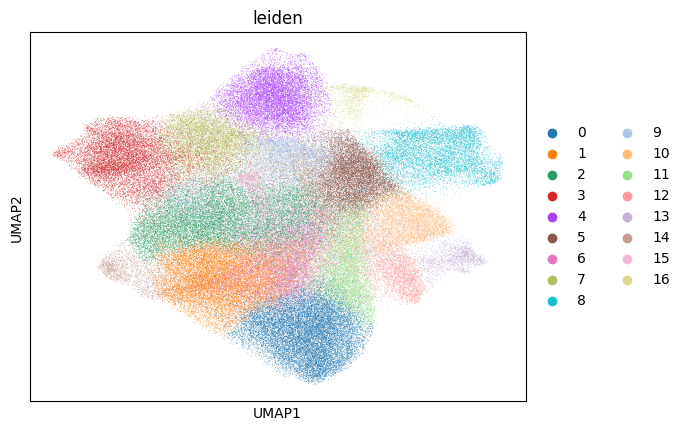
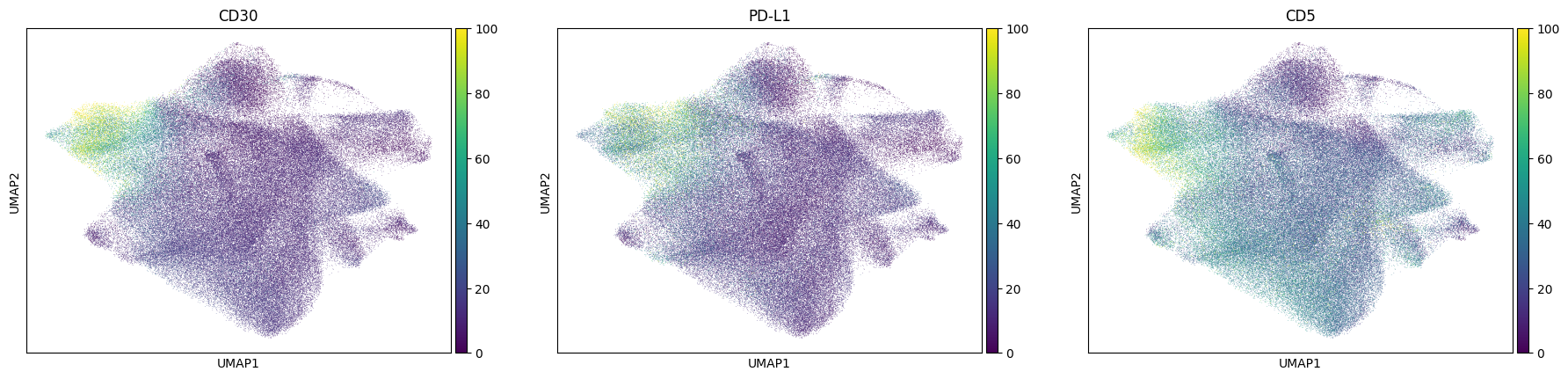

Next we try to estimate the measure of association between the KRONOS/Leiden clusters and the annotated cell type, by calculating the Cramers V statistic.
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import seaborn as sns
from scipy.stats import chi2_contingency
for _cluster_key in ["kronos", "leiden"]:
df = sdata[f"{table_name}_normalized"].obs
df = df[~df["cell_type_id"].isna()]
contingency = pd.crosstab(df["cell_type"], df[_cluster_key])
# chi squared test
chi2, p, dof, expected = chi2_contingency(contingency)
# cramers v
def _cramers_v(confusion_matrix):
chi2 = chi2_contingency(confusion_matrix)[0]
n = confusion_matrix.sum().sum()
phi2 = chi2 / n
r, k = confusion_matrix.shape
return np.sqrt(phi2 / min(k - 1, r - 1))
print("Cramers v:", _cramers_v(contingency))
print("Chi-squared statistic:", chi2)
print("p-value:", p)
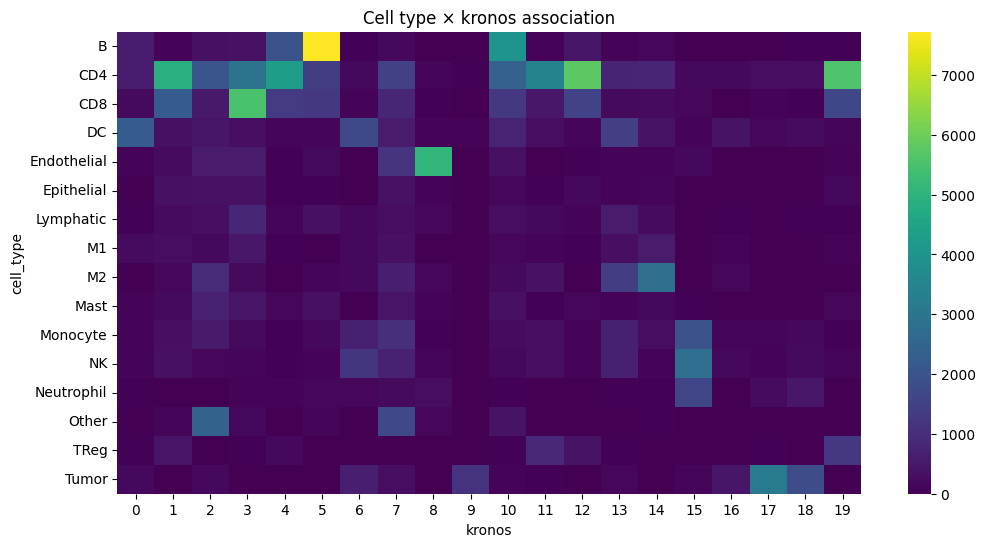
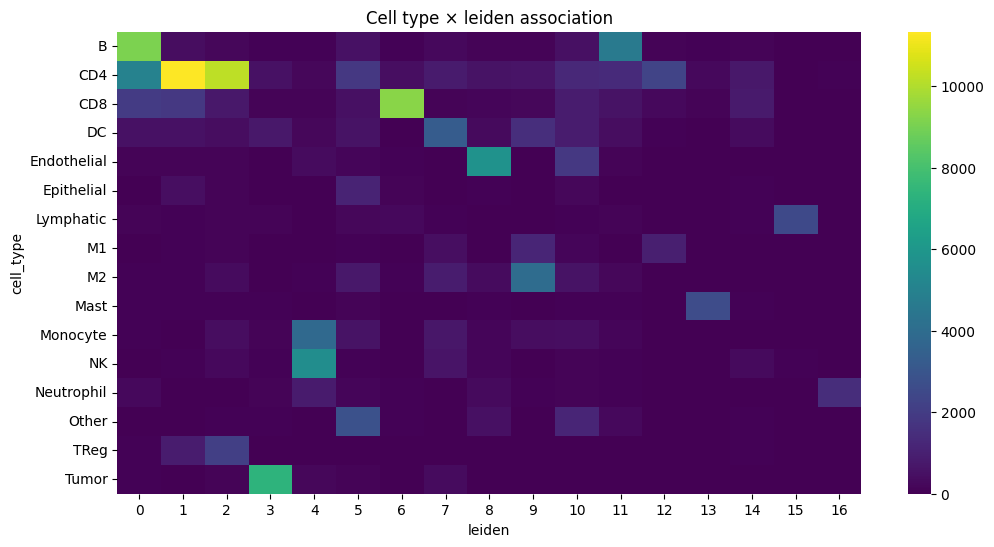
plt.figure(figsize=(12, 6))
sns.heatmap(contingency, cmap="viridis")
plt.title(f"Cell type × {_cluster_key} association")
plt.show()
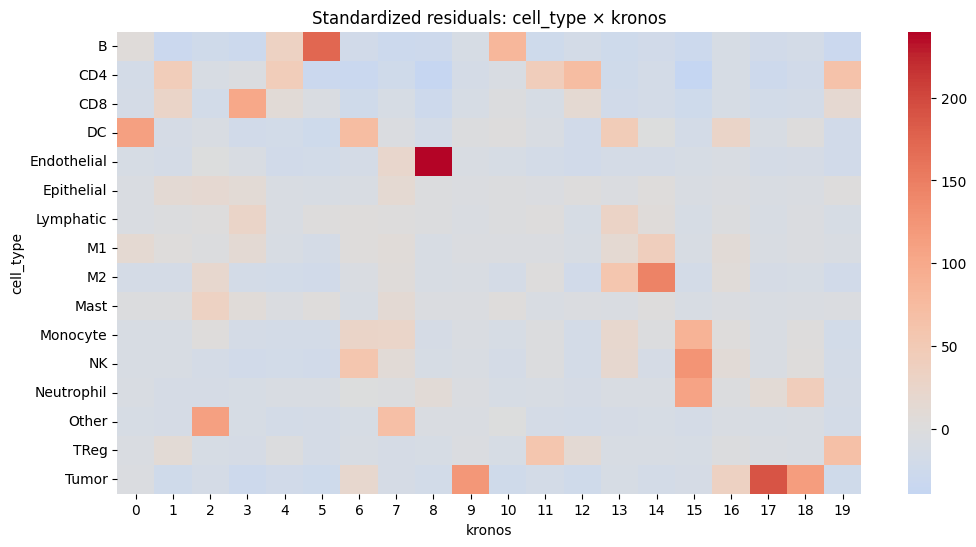
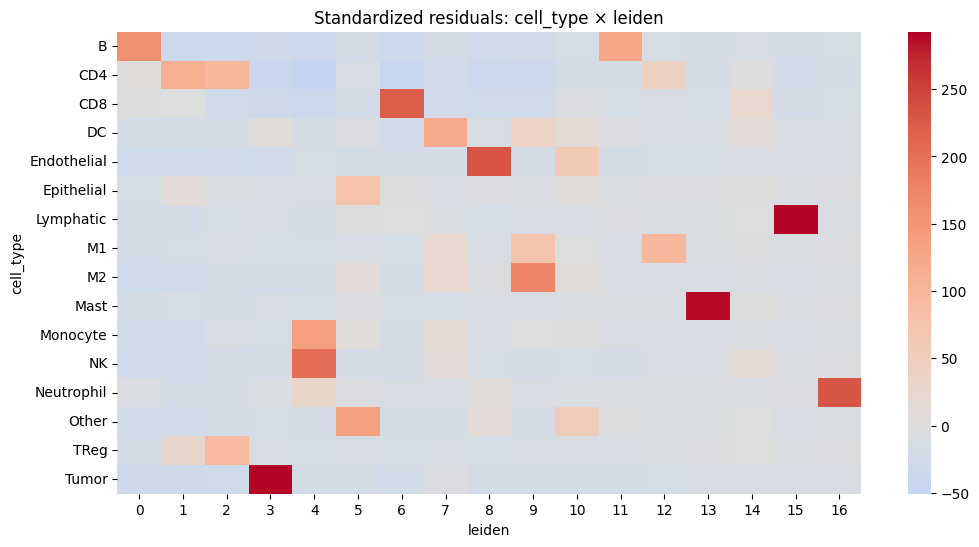
# Calculate standardized residuals
residuals = (contingency - expected) / np.sqrt(expected)
residuals_df = pd.DataFrame(residuals, index=contingency.index, columns=contingency.columns)
resid_long = residuals_df.stack().reset_index()
resid_long.columns = ["cell_type", _cluster_key, "std_residual"]
# Sort by strength of deviation
resid_long["abs_resid"] = resid_long["std_residual"].abs()
top_effects = resid_long.sort_values("abs_resid", ascending=False).head(20)
print(top_effects)
plt.figure(figsize=(12, 6))
sns.heatmap(residuals_df, center=0, cmap="coolwarm", annot=False)
plt.title(f"Standardized residuals: cell_type × {_cluster_key}")
plt.show()
Cramers v: 0.40833263281018034
Chi-squared statistic: 359473.4853451965
p-value: 0.0
cell_type kronos std_residual abs_resid
88 Endothelial 8 239.920615 239.920615
317 Tumor 17 190.376840 190.376840
5 B 5 173.119357 173.119357
174 M2 14 143.902390 143.902390
235 NK 15 125.539999 125.539999
309 Tumor 9 122.268829 122.268829
318 Tumor 18 114.545215 114.545215
262 Other 2 112.218924 112.218924
60 DC 0 110.584902 110.584902
255 Neutrophil 15 108.023549 108.023549
43 CD8 3 101.712499 101.712499
215 Monocyte 15 87.373576 87.373576
10 B 10 80.813422 80.813422
32 CD4 12 72.044409 72.044409
66 DC 6 72.007998 72.007998
267 Other 7 68.878832 68.878832
299 TReg 19 67.417829 67.417829
39 CD4 19 64.061963 64.061963
173 M2 13 57.987977 57.987977
226 NK 6 57.130258 57.130258
Cramers v: 0.573074536541328
Chi-squared statistic: 708045.0783542951
p-value: 0.0
cell_type leiden std_residual abs_resid
258 Tumor 3 292.654256 292.654256
117 Lymphatic 15 291.562718 291.562718
166 Mast 13 289.693060 289.693060
76 Endothelial 8 231.233794 231.233794
220 Neutrophil 16 228.920163 228.920163
40 CD8 6 222.583909 222.583909
191 NK 4 203.898082 203.898082
145 M2 9 175.362353 175.362353
0 B 0 160.610434 160.610434
174 Monocyte 4 139.437011 139.437011
226 Other 5 134.999515 134.999515
11 B 11 124.371550 124.371550
58 DC 7 122.938085 122.938085
18 CD4 1 112.172448 112.172448
19 CD4 2 98.468486 98.468486
131 M1 12 94.287696 94.287696
240 TReg 2 93.462668 93.462668
90 Epithelial 5 77.534145 77.534145
128 M1 9 74.155199 74.155199
78 Endothelial 10 59.498226 59.498226
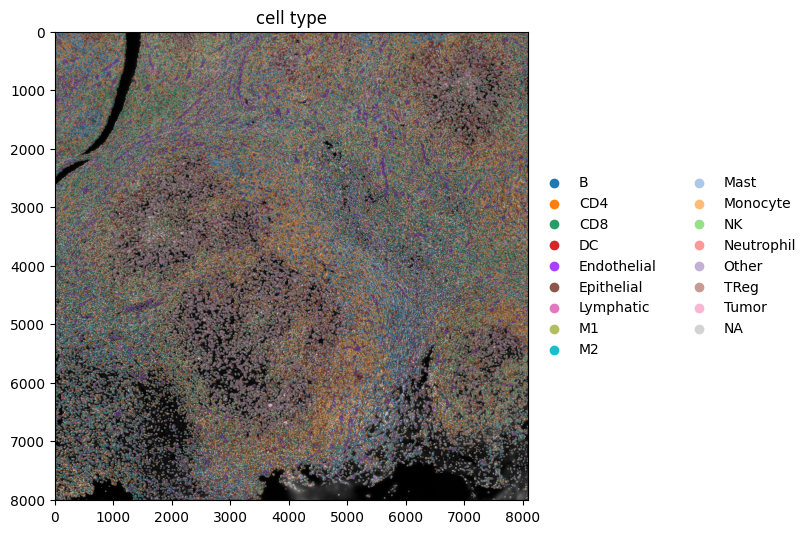
Next, we spatially plot the KRONOS and Leiden clusters, and the cell types.
import dask.array as da
import matplotlib.pyplot as plt
import numpy as np
from matplotlib.colors import Normalize
c = "DAPI-01"
table_name = f"{table_name}_normalized"
se = hp.im.get_dataarray(sdata, element_name="image")
channels = se.c.data
c_id = np.where(channels == c)[0].item()
vmax = da.percentile(se.data[c_id].flatten(), q=99).compute() # clip to 99% percentile
norm = Normalize(vmax=vmax, clip=True)
render_images_kwargs = {
"cmap": "grey",
"norm": norm,
"scale": "scale3",
}
render_labels_kwargs = {
"scale": "scale2",
"fill_alpha": 0.2,
"outline_alpha": 0.3,
}
show_kwargs = {"title": "kronos", "colorbar": False, "dpi": 100, "figsize": (8, 8)}
ax = hp.pl.plot_sdata(
sdata,
ax=None,
image_name="image",
channel=c,
labels_name="segmentation_mask",
table_name=table_name,
color="kronos",
render_images_kwargs=render_images_kwargs,
render_labels_kwargs=render_labels_kwargs,
show_kwargs=show_kwargs,
)
show_kwargs = {"title": "leiden", "colorbar": False, "dpi": 100, "figsize": (8, 8)}
ax = hp.pl.plot_sdata(
sdata,
ax=None,
image_name="image",
channel=c,
labels_name="segmentation_mask",
table_name=table_name,
color="leiden",
render_images_kwargs=render_images_kwargs,
render_labels_kwargs=render_labels_kwargs,
show_kwargs=show_kwargs,
)
show_kwargs = {"title": "cell type", "colorbar": False, "dpi": 100, "figsize": (8, 8)}
ax = hp.pl.plot_sdata(
sdata,
ax=None,
image_name="image",
channel=c,
labels_name="segmentation_mask",
table_name=table_name,
color="cell_type",
render_images_kwargs=render_images_kwargs,
render_labels_kwargs=render_labels_kwargs,
show_kwargs=show_kwargs,
)
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
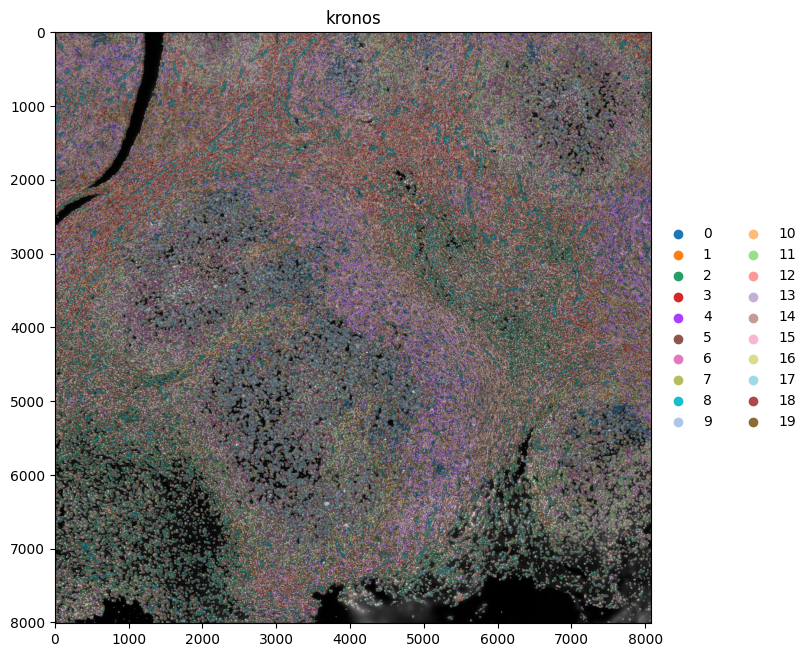
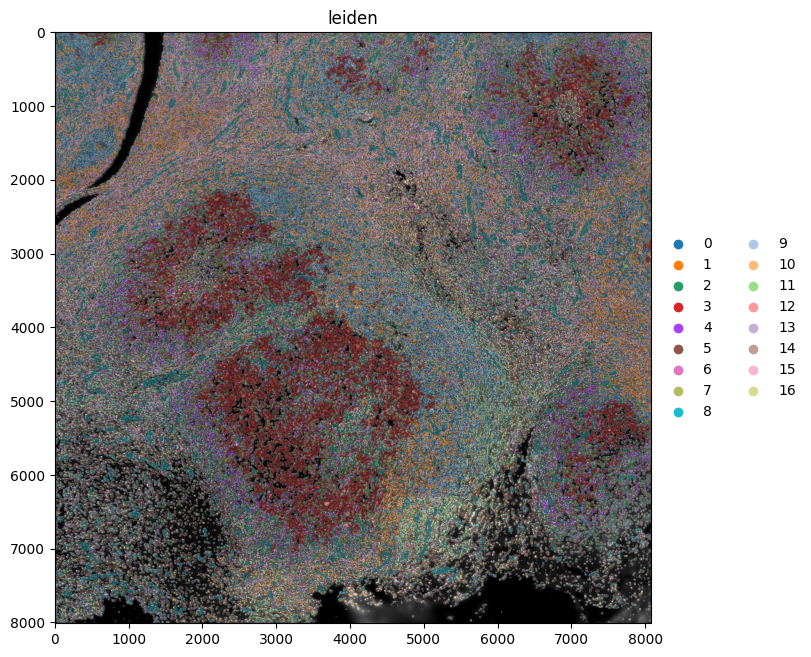
The tiling effects in the leiden clusters are caused by the tiling effects visible in some of the channels. By removing the channels “EGFR”, “FOXP3”, “PD-L1”, “T-BET”, “TCR-G-D”, “CD56” and “CD69”, a lot of the tiling effects were removed when spatially plotting the leiden clusters.
Note that when plotting the kronos clusters spatially, such tiling effects are not observed.
norm = Normalize(vmin=0, vmax=10000, clip=True)
render_images_kwargs = {
"cmap": "viridis",
"norm": norm,
"scale": "scale3",
}
for channel in [
"EGFR",
"FOXP3",
"PD-L1",
"T-BET",
"TCR-G-D",
"CD56",
"CD69",
]: # these channels causing tiling artefact in leiden clustering.
show_kwargs = {"title": str(channel), "colorbar": False, "dpi": 100, "figsize": (4, 4)}
ax = hp.pl.plot_sdata(
sdata,
ax=None,
image_name="image",
channel=channel,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
)
ax.axis("off")