Working with multiple samples (and coordinate systems)#
In this notebook, we will work with mouse liver spatial transcriptomics data generated using Resolve Biosciences’ Molecular Cartography technology, to illustrate the use of multiple samples and coordinate systems in Harpy and SpatialData.
1. Read the images into a SpatialData object backed by a zarr store.#
import os
import tempfile
import uuid
from spatialdata import SpatialData
import harpy as hp
sdata = SpatialData()
default_tmp_path = tempfile.gettempdir()
zarr_path = os.path.join(default_tmp_path, f"sdata_{uuid.uuid4()}.zarr")
sdata.write(zarr_path)
Add image elements.
from dask_image import imread
from spatialdata.transformations import Identity, Scale
from harpy.datasets.registry import get_registry
registry = get_registry()
arr_a1_1 = imread.imread(registry.fetch("transcriptomics/resolve/mouse/20272_slide1_A1-1_DAPI.tiff"))
arr_a1_2 = imread.imread(registry.fetch("transcriptomics/resolve/mouse/20272_slide1_A1-2_DAPI.tiff"))
pixel_size = 0.138 # pixel size in micron for resolve
scale = Scale(axes=["x", "y"], scale=[pixel_size, pixel_size])
sdata = hp.im.add_image(
sdata,
arr=arr_a1_1,
output_image_name="image_a1_1",
transformations={"a1_1_pixel": Identity(), "a1_1_micron": scale},
c_coords="DAPI",
scale_factors=[2, 2, 2, 2],
overwrite=True,
)
sdata = hp.im.add_image(
sdata,
arr=arr_a1_2,
output_image_name="image_a1_2",
transformations={"a1_2_pixel": Identity(), "a1_2_micron": scale},
c_coords="DAPI",
scale_factors=[2, 2, 2, 2],
overwrite=True,
)
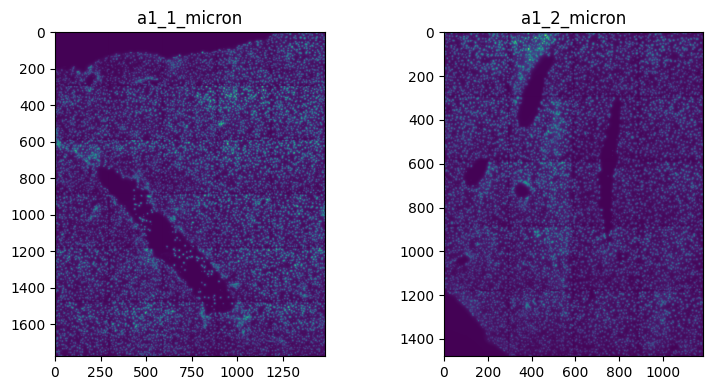
2. Specify coordinate system when plotting.#
import matplotlib.pyplot as plt
fig, axes = plt.subplots(1, 2, figsize=(8, 4))
channel = "DAPI"
render_images_kwargs = {
"cmap": "viridis",
}
image_name = ["image_a1_1", "image_a1_2"]
to_coordinate_system = ["a1_1_micron", "a1_2_micron"]
for _image_name, _to_coordinate_system, _ax in zip(image_name, to_coordinate_system, axes, strict=True):
show_kwargs = {"title": _to_coordinate_system, "colorbar": False}
_ax = hp.pl.plot_sdata(
sdata,
image_name=_image_name,
channel=channel,
crd=None,
to_coordinate_system=_to_coordinate_system,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=_ax,
)
plt.tight_layout()
plt.show()
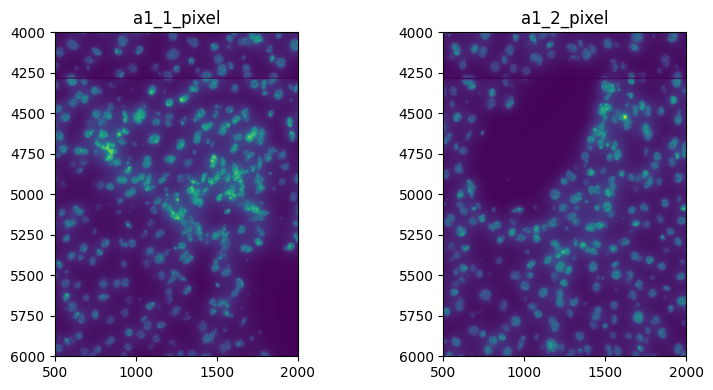
Visualize a crop:
import matplotlib.pyplot as plt
fig, axes = plt.subplots(1, 2, figsize=(8, 4))
channel = "DAPI"
render_images_kwargs = {
"cmap": "viridis",
}
image_name = ["image_a1_1", "image_a1_2"]
to_coordinate_system = ["a1_1_pixel", "a1_2_pixel"]
for _image_name, _to_coordinate_system, _ax in zip(image_name, to_coordinate_system, axes, strict=True):
show_kwargs = {"title": _to_coordinate_system, "colorbar": False}
_ax = hp.pl.plot_sdata(
sdata,
image_name=_image_name,
channel=channel,
crd=[500, 2000, 4000, 6000],
to_coordinate_system=_to_coordinate_system,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=_ax,
)
plt.tight_layout()
plt.show()
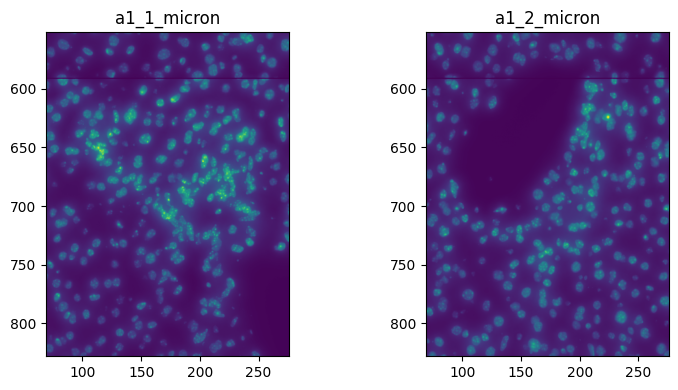
Or in micron
import matplotlib.pyplot as plt
fig, axes = plt.subplots(1, 2, figsize=(8, 4))
channel = "DAPI"
render_images_kwargs = {
"cmap": "viridis",
}
image_name = ["image_a1_1", "image_a1_2"]
to_coordinate_system = ["a1_1_micron", "a1_2_micron"]
for _image_name, _to_coordinate_system, _ax in zip(image_name, to_coordinate_system, axes, strict=True):
show_kwargs = {"title": _to_coordinate_system, "colorbar": False}
_ax = hp.pl.plot_sdata(
sdata,
image_name=_image_name,
channel=channel,
crd=[69, 276, 552, 828],
to_coordinate_system=_to_coordinate_system,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=_ax,
)
plt.tight_layout()
plt.show()
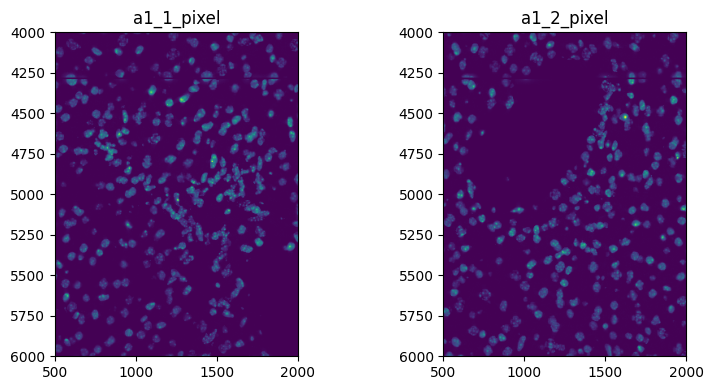
3. Image processing.#
Do some image processing on the full image, no need to specify a coordinate system
sdata = hp.im.min_max_filtering(
sdata, image_name="image_a1_1", output_image_name="image_a1_1_min_max", size_min_max_filter=40, overwrite=True
)
When doing some image processing on a crop, you need to specify the coordinate system to which the crop is defined
for image_name, to_coordinate_system in zip(["image_a1_1", "image_a1_2"], ["a1_1_pixel", "a1_2_pixel"], strict=True):
sdata = hp.im.min_max_filtering(
sdata,
image_name=image_name,
output_image_name=f"{image_name}_min_max",
size_min_max_filter=40,
crd=[500, 2000, 4000, 6000],
to_coordinate_system=to_coordinate_system,
overwrite=True,
)
fig, axes = plt.subplots(1, 2, figsize=(8, 4))
channel = "DAPI"
render_images_kwargs = {
"cmap": "viridis",
}
for image_name, to_coordinate_system, ax in zip(
["image_a1_1_min_max", "image_a1_2_min_max"], ["a1_1_pixel", "a1_2_pixel"], axes, strict=True
):
show_kwargs = {"title": to_coordinate_system, "colorbar": False}
ax = hp.pl.plot_sdata(
sdata,
image_name=image_name,
channel=channel,
crd=None,
to_coordinate_system=to_coordinate_system,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=ax,
)
plt.tight_layout()
plt.show()
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
4. Segmentation.#
# no need to specify coordinate system if no crop is specifed
import torch
from dask.distributed import LocalCluster, Client
cluster = LocalCluster(
n_workers=1
if torch.backends.mps.is_available() or torch.cuda.is_available()
else 8, # If mps/cuda device available, it is better to increase chunk size to maximal value that fits on the gpu, and set n_workers to 1.
threads_per_worker=1,
memory_limit="32GB",
# local_directory=os.environ.get( "TMPDIR" ),
)
client = Client(cluster)
print(client.dashboard_link)
sdata = hp.im.segment(
sdata,
image_name="image_a1_1_min_max",
output_labels_name="labels_a1_1",
output_shapes_name="shapes_a1_1",
overwrite=True,
crd=None,
)
# but need to specify coordinate system when crop is defined
sdata = hp.im.segment(
sdata,
image_name="image_a1_2_min_max",
output_labels_name="labels_a1_2",
output_shapes_name="shapes_a1_2",
overwrite=True,
crd=[600, 2000, 4000, 6000],
to_coordinate_system="a1_2_pixel",
)
client.close()
http://127.0.0.1:64440/status
fig, axes = plt.subplots(1, 2, figsize=(8, 4))
channel = "DAPI"
image_name = "image_a1_2_min_max"
labels_name = "labels_a1_2"
crd = [600, 2000, 4000, 6000] # Crop in pixels. One could also choose to do the crop in micron coordinates
to_coordinate_system = "a1_2_pixel"
render_images_kwargs = {"cmap": "viridis"}
render_labels_kwargs = {"fill_alpha": 0.6, "outline_alpha": 0.4}
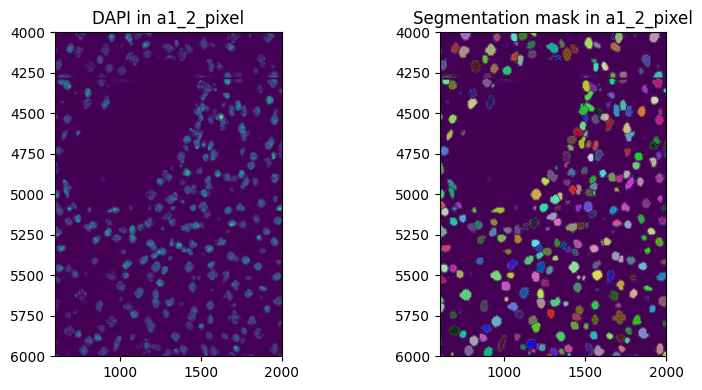
show_kwargs = {"title": f"DAPI in {to_coordinate_system}", "colorbar": False}
_ax = hp.pl.plot_sdata(
sdata,
image_name=image_name,
crd=crd,
to_coordinate_system=to_coordinate_system,
channel=channel,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=axes[0],
)
# _ax.axis("off")
show_kwargs = {"title": f"Segmentation mask in {to_coordinate_system}", "colorbar": False}
_ax = hp.pl.plot_sdata(
sdata,
image_name=image_name,
labels_name=labels_name,
crd=crd,
to_coordinate_system=to_coordinate_system,
channel=channel,
render_images_kwargs=render_images_kwargs,
render_labels_kwargs=render_labels_kwargs,
show_kwargs=show_kwargs,
ax=axes[1],
)
# _ax.axis("off")
plt.tight_layout()
plt.show()
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
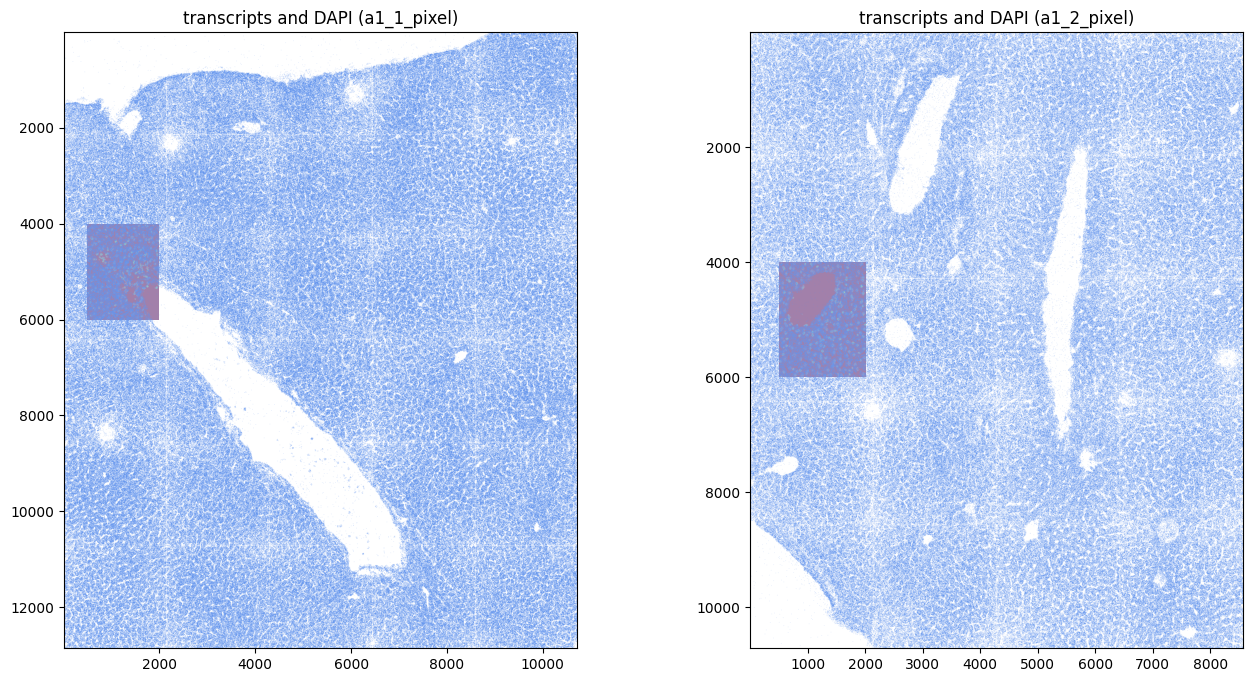
Read the transcripts downloaded previously. For downstream analysis it is convenient if the transcripts are stored in pixels (intrinsic coordinate system of the images), and registered with the images. Therefore, if transcripts are delivered in micron, it is possible to specify an affine transformation via the parameter transform_matrix of the function harpy.io.read_transcripts that converts from micron to pixels, and registers the transcripts with the images. A micron coordinate system will be added to the resulting points element of the SpatialData object if to_micron_coordinate_system is specified. We refer to the docstring of harpy.io.read_transcripts for more information.
Transcripts from Resolve Biosciences’ Molecular Cartography technology are already in pixels. By specifying the pixel size and the micron coordinate system, the function harpy.io.read_transcripts takes care of adding a micron coordinate system.
path_transcripts_a1_1 = registry.fetch("transcriptomics/resolve/mouse/20272_slide1_A1-1_results.txt")
path_transcripts_a1_2 = registry.fetch("transcriptomics/resolve/mouse/20272_slide1_A1-2_results.txt")
sdata = hp.io.read_transcripts(
sdata,
path_count_matrix=path_transcripts_a1_1,
column_x=0,
column_y=1,
column_gene=3,
delimiter="\t",
header=None,
output_points_name="transcripts_a1_1",
to_coordinate_system="a1_1_pixel",
to_micron_coordinate_system="a1_1_micron",
pixel_size=pixel_size, # size of pixels in micron
transform_matrix=None, # transcripts are already in pixels, and registered with the images
overwrite=True,
)
sdata = hp.io.read_transcripts(
sdata,
path_count_matrix=path_transcripts_a1_2,
column_x=0,
column_y=1,
column_gene=3,
delimiter="\t",
header=None,
output_points_name="transcripts_a1_2",
to_coordinate_system="a1_2_pixel",
to_micron_coordinate_system="a1_2_micron",
pixel_size=pixel_size, # size of pixels in micron
transform_matrix=None, # transcripts are already in pixels, and registered with the images
overwrite=True,
)
Verify that transformation associated to points element in the pixel coordinate system is indeed the identity, and verify that the labels have same transformation associated to them.
from spatialdata.transformations import get_transformation
print(
f"points:\n {get_transformation(sdata['transcripts_a1_2'], to_coordinate_system='a1_2_pixel')}\n",
)
print(
f"image:\n{get_transformation(sdata['image_a1_2_min_max'], to_coordinate_system='a1_2_pixel')}\n",
) # one crop was taken, so an extra translation is defined on image.
print(
f"image:\n{get_transformation(sdata['labels_a1_2'], to_coordinate_system='a1_2_pixel')}\n",
) # two times a crop was taken, so a sequence of two translations is defined on labels and the associated shapes.
print(
f"shapes:\n{get_transformation(sdata['shapes_a1_2'], to_coordinate_system='a1_2_pixel')}\n",
) # two times a crop was taken, so a sequence of two translations is defined on labels and the associated shapes.
points:
Identity
image:
Sequence
Translation (c, y, x)
[ 0. 4000. 500.]
Identity
image:
Sequence
Translation (y, x)
[ 0. 100.]
Sequence
Translation (y, x)
[4000. 500.]
Identity
shapes:
Sequence
Translation (x, y)
[100. 0.]
Sequence
Translation (x, y)
[ 500. 4000.]
Identity
fig, axes = plt.subplots(1, 2, figsize=(16, 8))
render_images_kwargs = {
"cmap": "viridis",
"alpha": 0.5,
}
image_name = ["image_a1_1_min_max", "image_a1_2_min_max"]
points_name = ["transcripts_a1_1", "transcripts_a1_2"]
to_coordinate_system = ["a1_1_pixel", "a1_2_pixel"]
for _image_name, _points_name, _to_coordinate_system, _ax in zip(
image_name, points_name, to_coordinate_system, axes, strict=True
):
show_kwargs = {
"title": f"transcripts and DAPI ({_to_coordinate_system})",
"colorbar": False,
}
hp.pl.plot_sdata_genes(
sdata,
points_name=_points_name,
image_name=_image_name,
genes=None, # plot all genes
color="cornflowerblue",
size=0.1,
frac=0.5, # only plot half of them
to_coordinate_system=_to_coordinate_system,
show_kwargs=show_kwargs,
render_images_kwargs=render_images_kwargs,
ax=_ax,
)
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Rasterizing image for faster rendering.
INFO Using 'datashader' backend with 'None' as reduction method to speed up plotting. Depending on the
reduction method, the value range of the plot might change. Set method to 'matplotlib' do disable this
behaviour.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Rasterizing image for faster rendering.
INFO Using 'datashader' backend with 'None' as reduction method to speed up plotting. Depending on the
reduction method, the value range of the plot might change. Set method to 'matplotlib' do disable this
behaviour.
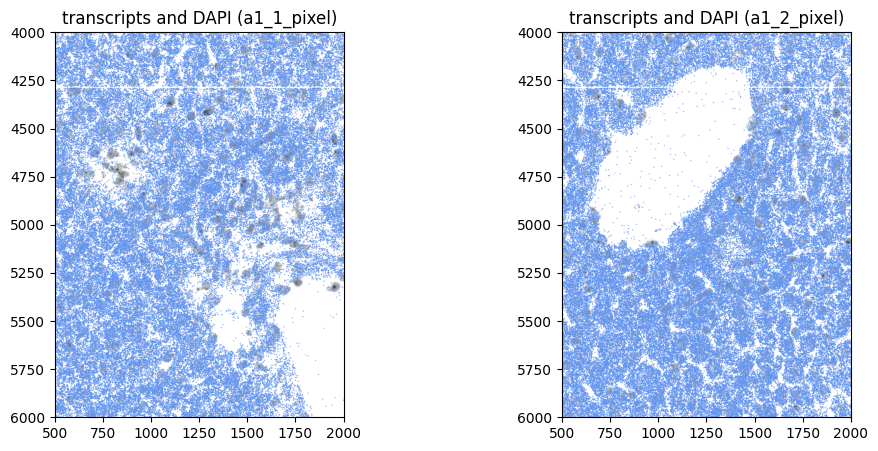
Or visualize a crop:
# we query the points elements outside of spatialdata, because querying points via spatialdata.bounding_box_query is slow
crd = [500, 2000, 4000, 6000]
points_name = ["transcripts_a1_1", "transcripts_a1_2"]
to_coordinate_system = ["a1_1_pixel", "a1_2_pixel"]
for _points_name, _to_coordinate_system in zip(points_name, to_coordinate_system, strict=True):
to_coordinate_system = _to_coordinate_system # we query in 'intrinsic' pixel coordinate system
y_start = crd[2]
x_start = crd[0]
y_end = crd[3]
x_end = crd[1]
name_x = "x"
name_y = "y"
y_query = f"{y_start} <={name_y} < {y_end}"
x_query = f"{x_start} <={name_x} < {x_end}"
query = f"{y_query} and {x_query}"
sdata[f"{_points_name}_queried"] = sdata[_points_name].query(query)
fig, axes = plt.subplots(1, 2, figsize=(12, 5))
render_images_kwargs = {
"cmap": "binary",
}
image_name = ["image_a1_1_min_max", "image_a1_2_min_max"]
points_name = [f"{_points_name}_queried" for _points_name in points_name]
to_coordinate_system = ["a1_1_pixel", "a1_2_pixel"]
for _image_name, _points_name, _to_coordinate_system, _ax in zip(
image_name, points_name, to_coordinate_system, axes, strict=True
):
show_kwargs = {
"title": f"transcripts and DAPI ({_to_coordinate_system})",
"colorbar": False,
}
hp.pl.plot_sdata_genes(
sdata,
points_name=_points_name,
image_name=_image_name,
genes=None, # plot all genes
color="cornflowerblue",
size=1,
frac=None, # only plot half of them
to_coordinate_system=_to_coordinate_system,
show_kwargs=show_kwargs,
render_images_kwargs=render_images_kwargs,
ax=_ax,
)
del sdata[_points_name]
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Rasterizing image for faster rendering.
INFO Using 'datashader' backend with 'None' as reduction method to speed up plotting. Depending on the
reduction method, the value range of the plot might change. Set method to 'matplotlib' do disable this
behaviour.
INFO Value for parameter 'color' appears to be a color, using it as such.
INFO Rasterizing image for faster rendering.
INFO Using 'datashader' backend with 'None' as reduction method to speed up plotting. Depending on the
reduction method, the value range of the plot might change. Set method to 'matplotlib' do disable this
behaviour.
5. Create the AnnData table.#
# Use dask cluster for optimal processing. For this dummy example, one could also work without a dask cluster.
cluster = LocalCluster(
n_workers=8,
threads_per_worker=1,
processes=True,
memory_limit="8GB",
# local_directory=os.environ.get( "TMPDIR" ),
)
client = Client(cluster)
print(client.dashboard_link)
sdata = hp.tb.allocate(
sdata,
labels_name="labels_a1_1",
points_name="transcripts_a1_1",
to_coordinate_system="a1_1_pixel", # allocation step should always be run in pixel coordinates
output_table_name="table",
update_shapes_elements=False,
append=False,
overwrite=True,
)
# append gene count of labels_a1_2 and points_a1_2 to anndata object with name 'table'
sdata = hp.tb.allocate(
sdata,
labels_name="labels_a1_2",
points_name="transcripts_a1_2",
to_coordinate_system="a1_2_pixel",
output_table_name="table",
update_shapes_elements=False,
append=True,
overwrite=True,
)
client.close()
http://127.0.0.1:64454/status
from spatialdata.models import TableModel
print(sdata["table"].uns[TableModel.ATTRS_KEY], "\n") # -> the table is annotated by "labels_a1_1" and "labels_a1_2"
print(
sdata["table"].uns[TableModel.ATTRS_KEY][TableModel.REGION_KEY_KEY], "\n"
) # -> column in .obs that holds the spatial element that is annotating the instance
print(
sdata["table"].uns[TableModel.ATTRS_KEY][TableModel.INSTANCE_KEY]
) # -> column in .obs that holds the instance id in the corresponding spatial element
{'region': ['labels_a1_1', 'labels_a1_2'], 'region_key': 'fov_labels', 'instance_key': 'cell_ID'}
fov_labels
cell_ID
display(sdata["table"].obs.head())
display(sdata["table"].obs.tail())
| cell_ID | fov_labels | |
|---|---|---|
| cells | ||
| 18_labels_a1_1_7f8836a8 | 18 | labels_a1_1 |
| 19_labels_a1_1_7f8836a8 | 19 | labels_a1_1 |
| 20_labels_a1_1_7f8836a8 | 20 | labels_a1_1 |
| 21_labels_a1_1_7f8836a8 | 21 | labels_a1_1 |
| 22_labels_a1_1_7f8836a8 | 22 | labels_a1_1 |
| cell_ID | fov_labels | |
|---|---|---|
| cells | ||
| 346_labels_a1_2_caa1aa78 | 346 | labels_a1_2 |
| 348_labels_a1_2_caa1aa78 | 348 | labels_a1_2 |
| 349_labels_a1_2_caa1aa78 | 349 | labels_a1_2 |
| 352_labels_a1_2_caa1aa78 | 352 | labels_a1_2 |
| 353_labels_a1_2_caa1aa78 | 353 | labels_a1_2 |
6. Leiden clustering.#
sdata = hp.tb.preprocess_transcriptomics(
sdata,
labels_name=["labels_a1_1", "labels_a1_2"],
table_name="table",
output_table_name="table_preprocessed",
update_shapes_elements=False,
overwrite=True,
) # we can also choose to set output_table_name equal to 'table'.
hp.tb.leiden(
sdata,
labels_name=["labels_a1_1", "labels_a1_2"],
table_name="table_preprocessed",
output_table_name="table_preprocessed",
overwrite=True,
)
SpatialData object, with associated Zarr store: /private/var/folders/q5/7yhs0l6d0x771g7qdbhvkvmr0000gp/T/sdata_66a1e781-b62b-463e-b4be-ef244dc695ba.zarr
├── Images
│ ├── 'image_a1_1': DataTree[cyx] (1, 12864, 10720), (1, 6432, 5360), (1, 3216, 2680), (1, 1608, 1340), (1, 804, 670)
│ ├── 'image_a1_1_min_max': DataArray[cyx] (1, 2000, 1500)
│ ├── 'image_a1_2': DataTree[cyx] (1, 10720, 8576), (1, 5360, 4288), (1, 2680, 2144), (1, 1340, 1072), (1, 670, 536)
│ └── 'image_a1_2_min_max': DataArray[cyx] (1, 2000, 1500)
├── Labels
│ ├── 'labels_a1_1': DataArray[yx] (2000, 1500)
│ └── 'labels_a1_2': DataArray[yx] (2000, 1400)
├── Points
│ ├── 'transcripts_a1_1': DataFrame with shape: (<Delayed>, 3) (2D points)
│ └── 'transcripts_a1_2': DataFrame with shape: (<Delayed>, 3) (2D points)
├── Shapes
│ ├── 'shapes_a1_1': GeoDataFrame shape: (341, 1) (2D shapes)
│ └── 'shapes_a1_2': GeoDataFrame shape: (295, 1) (2D shapes)
└── Tables
├── 'table': AnnData (630, 88)
└── 'table_preprocessed': AnnData (605, 78)
with coordinate systems:
▸ 'a1_1_micron', with elements:
image_a1_1 (Images), image_a1_1_min_max (Images), labels_a1_1 (Labels), transcripts_a1_1 (Points), shapes_a1_1 (Shapes)
▸ 'a1_1_pixel', with elements:
image_a1_1 (Images), image_a1_1_min_max (Images), labels_a1_1 (Labels), transcripts_a1_1 (Points), shapes_a1_1 (Shapes)
▸ 'a1_2_micron', with elements:
image_a1_2 (Images), image_a1_2_min_max (Images), labels_a1_2 (Labels), transcripts_a1_2 (Points), shapes_a1_2 (Shapes)
▸ 'a1_2_pixel', with elements:
image_a1_2 (Images), image_a1_2_min_max (Images), labels_a1_2 (Labels), transcripts_a1_2 (Points), shapes_a1_2 (Shapes)
fig, axes = plt.subplots(1, 2, figsize=(12, 5))
channel = "DAPI"
image_name = ["image_a1_1_min_max", "image_a1_2_min_max"]
labels_name = ["labels_a1_1", "labels_a1_2"]
to_coordinate_system = ["a1_1_pixel", "a1_2_pixel"]
table_name = "table_preprocessed"
crd = [600, 2000, 4000, 6000]
render_images_kwargs = {"cmap": "binary"}
render_labels_kwargs = {"fill_alpha": 0.6, "outline_alpha": 0.4}
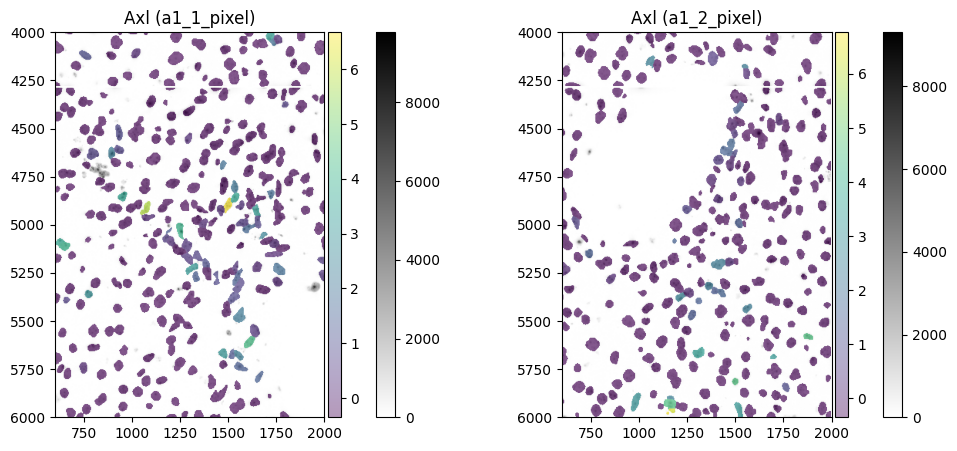
color = "Axl"
for _image_name, _labels_name, _to_coordinate_system, _ax in zip(
image_name, labels_name, to_coordinate_system, axes, strict=True
):
# subset because querying of points is slow, and we do not need them in this visualization
sdata_to_plot = sdata.subset(element_names=[_image_name, _labels_name, table_name])
show_kwargs = {
"title": f"{color} ({_to_coordinate_system})",
"colorbar": True,
}
_ax = hp.pl.plot_sdata(
sdata_to_plot,
image_name=_image_name,
channel=channel,
crd=crd,
to_coordinate_system=_to_coordinate_system,
labels_name=_labels_name,
table_name=table_name,
color=color,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=_ax,
)
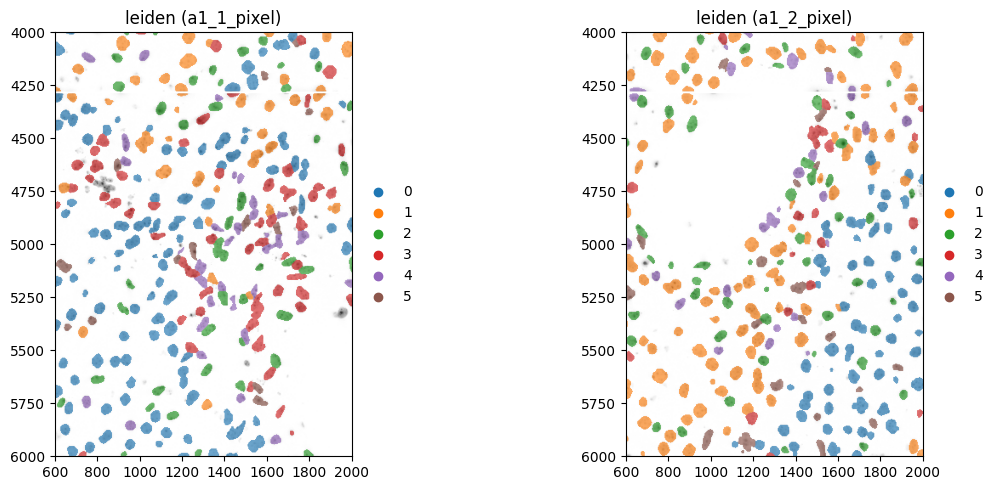
fig, axes = plt.subplots(1, 2, figsize=(12, 5))
color = "leiden"
for _image_name, _labels_name, _to_coordinate_system, _ax in zip(
image_name, labels_name, to_coordinate_system, axes, strict=True
):
sdata_to_plot = sdata.subset(element_names=[_image_name, _labels_name, table_name])
show_kwargs = {
"title": f"{color} ({_to_coordinate_system})",
"colorbar": False,
}
_ax = hp.pl.plot_sdata(
sdata_to_plot,
image_name=_image_name,
channel=channel,
crd=crd,
to_coordinate_system=_to_coordinate_system,
labels_name=_labels_name,
table_name=table_name,
color=color,
render_images_kwargs=render_images_kwargs,
show_kwargs=show_kwargs,
ax=_ax,
)
plt.tight_layout()
plt.show()
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
INFO Rasterizing image for faster rendering.
7. Query the labels elements and the associated table element it annotates.#
zarr_path = os.path.join(default_tmp_path, f"sdata_{uuid.uuid4()}.zarr") # output path for queried sdata
labels_name = [
"labels_a1_1",
"labels_a1_2",
]
crd = [
(600, 1000, 4000, 5000),
(700, 1000, 4000, 5000),
] # queries could also be done in pixels
to_coordinate_system = ["a1_1_pixel", "a1_2_pixel"]
sdata_queried = hp.utils.bounding_box_query(
sdata,
labels_name=labels_name,
crd=crd,
to_coordinate_system=to_coordinate_system,
copy_image=False,
copy_shapes=False,
copy_points=False,
output=zarr_path,
)
for _labels_name in labels_name:
assert _labels_name in sdata_queried.labels
from spatialdata.transformations import get_transformation
get_transformation(sdata_queried["labels_a1_1"], to_coordinate_system="a1_1_pixel")
Sequence
Translation (y, x)
[ 0. 100.]
Sequence
Translation (y, x)
[4000. 500.]
Identity
sdata_queried["table_preprocessed"]
AnnData object with n_obs × n_vars = 66 × 78
obs: 'cell_ID', 'fov_labels', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_2_genes', 'pct_counts_in_top_5_genes', 'n_counts', 'shapeSize', 'leiden'
var: 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'n_cells', 'mean', 'std'
uns: 'pca', 'spatialdata_attrs', 'umap', 'leiden', 'rank_genes_groups', 'log1p', 'neighbors'
obsm: 'X_pca', 'X_umap', 'spatial'
varm: 'PCs'
layers: 'raw_counts'
obsp: 'connectivities', 'distances'